Letters (Selected Paper)
Molecular Dynamics Simulation of Structure Change of β-LiAlSiO4 with Temperature
2020 Volume 19 Issue 4 Pages 146-148
Details
2020 Volume 19 Issue 4 Pages 146-148
Anisotropic low-thermal expansion of β-LiAlSiO4 crystal was reproduced by MD calculation applying a potential model of a spherically symmetric two-body interatomic interaction. From the correlation between the Si-O-Al angle and the Si-Al distance in the c-axis direction, the expansion of a-axis and the contract of c-axis brings the split of Si-O-Al angle distribution. At the same time, the stable position of Li+ ion sites also changed. This suggests the cation position also important in the mechanism of low-thermal expansion observed in silicate materials.
調理器具や集積回路の基板のように作動時に高温に達する製品や部品には,熱膨張・収縮の繰り返しによる破損や劣化を防ぐ工夫が必要である.方策の一つとして基板に用いることができる熱膨張の低い材料が求められている.β-LiAlSiO4は実用されている低熱膨張材料である.β-LiAlSiO4の低熱膨張は,昇温に伴い六方晶のa軸が伸張しc軸が収縮する異方性の熱膨張特性に依拠することが分かっているが,新しい低熱膨張材料を見出すためには機構をより詳細に知る必要があると考え,分子動力学(MD)法による検討を進めてきた.これまでにMD法でβ-LiAlSiO4の低熱膨張の特性を再現可能であることを確認し,機構の検討について報告を行った [1].本稿ではβ-LiAlSiO4結晶単位格子の温度変化がLi+イオンの配置へ及ぼす影響を報告する.
本研究のMD計算には,(1)式に示す2体間原子間相互作用ポテンシャル関数を用いた [2].β-LiAlSiO4結晶
β-LiAlSiO4はSiO4-,AlO4-四面体ユニットが酸化物イオンを介して交互に結合して形成する二重らせんがc軸方向へ連なる構造を有している(Figure 1).Figure 2 に昇温に伴う格子定数a, cの変化を示す.原子間相互作用の調整により,MD計算の結果(〇)は実測値(×)と±0.6% 以内で一致し,昇温に従いa軸が伸び,c軸が縮む傾向を再現している.昇温に伴うa軸の伸張はらせんの半径が拡がることを意味する.同時にc軸が縮むため,体積膨張は抑制される.MD計算から得られた線熱膨張係数をTable 1に示す.
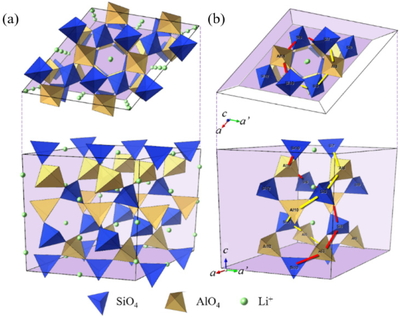
Unit cell of β-LiAlSiO4 (a) and the intrinsic double spiral structure of this crystal (b).
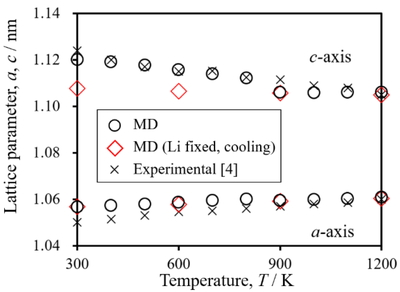
Lattice parameter, a and c of β-LiAlSiO4.
| ΔaL ppm / K−1 | ΔcL ppm / K−1 | |
| MD (This work) | 4.35 | −14.2 |
| Experimental [4] | 8.6 | −18.4 |
MD計算より得られたSi-OとAl-Oの2体相関関数から,SiO4とAlO4ユニットを構成するSi-O結合とAl-O結合の距離は温度で変化せずほぼ一定であった.一方,SiO4とAlO4を繋ぐSi-O-Al結合角θと,隣接四面体間の距離のc軸に平行な成分
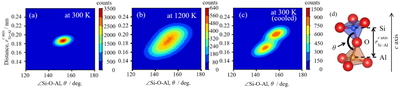
Relation between ∠Si-O-Al angle and Si-Al distance (see (d)) at 300 K (a) and at 1200 K (b). (c) is a result of cooling MD simulation from 1200 K to 300 K under condition that Li+ relative positions were fixed artificially.
Li+サイトは二重らせんの中心にある(Figure 1).また,c軸方向へ電場を印加したMD計算でLi+がc軸方向へ移動することがわかっている [1].Li+イオンの分布位置の温度依存性を調べた結果をFigure 4に示す.Li+イオンは300 Kでは隣接する四面体のSiあるいはAl原子の横にあり,600 Kまでほぼそのままであった.しかし,900 K 以上ではLi+イオンはSiとAlの横から移動した.すなわち,Figure 4中に示したLi-O間距離のc軸方向成分を座標とすると,Oの横(座標ゼロ)に,Li+イオンの新たな安定サイトが生じることが示された.
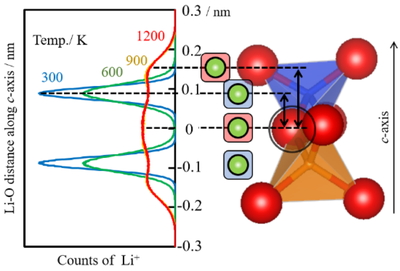
Distribution of Li+ ion position.
球対称の2体間原子間相互作用系のポテンシャルモデルをβ-LiAlSiO4結晶へ適用したMD計算により,異方性の低熱膨張が再現された.シリケート系の低熱膨張材料に対して提案されている機構モデル [5, 6]では陽イオンの安定サイトの変化までは考慮されておらず,今後同様の検討が必要であると考えられる.