Abstract
Density functional theory (DFT) is a successful theory for calculating the electronic structure of atoms, molecules, and solids. However, in modern computing environments is difficult significantly improve the computational efficiency of DFT. Acceleration of DFT requires optimization of the computational algorithms. We demonstrate two acceleration methods by optimization of the computational algorithms that optimize parallel parameters for eigenvalue calculations and optimization of convergence conditions for the self-consistent field (SCF) calculation.
Translated Abstract
Density functional theory (DFT) is a successful theory for calculating the electronic structure of atoms, molecules, and solids. However, in modern computing environments is difficult significantly improve the computational efficiency of DFT. Acceleration of DFT requires optimization of the computational algorithms. We demonstrate two acceleration methods by optimization of the computational algorithms that optimize parallel parameters for eigenvalue calculations and optimization of convergence conditions for the self-consistent field (SCF) calculation.
1 Introduction
近年, 材料開発においても研究開発期間の短縮, 低コスト化を目指すデータ駆動型研究開発の取り組みとして, Materials Informatics (MI)が世界的に進展している [1]. MIは良質な材料データの蓄積が重要となり, 実験に加えて計算化学などによる効率的なデータ蓄積が期待される. 計算化学の主要な計算手法である密度汎関数法(Density Functional Theory, DFT)は, 既存の計算環境における大幅な計算効率の向上は困難であり, 計算化学の高速化にはプロセッサアーキテクチャ, 専用回路, 計算アルゴリズムの最適化を含めた検討が必要である [2, 3].
本研究では, DFTの自己無撞着(Self-Consistent Field, SCF)な計算において, 大規模な系では主要な計算コストの一つである固有値計算の並列条件の最適化, SCF計算の繰返し回数を決定する収束条件の最適化, この2つの計算アルゴリズムの最適化による高速化手法を示す.
2 Proposal
2.1 Optimization of eigenvalue calculation
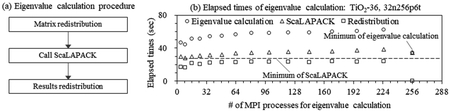
固有値計算の並列条件の最適化について説明する. Figure 1aはSCF計算における固有値計算の手順を示す. 計算化学ソフトウェアではDFT全体と固有値計算の並列数は異なっても良い. 固有値の計算はScaLAPACKなどの数学ライブラリを呼出して計算し, DFT全体と固有値計算の並列数が異なる場合, 行列, 計算結果の再配布が必要である. Figure 1bはTiO2 36分子モデルの固有値計算, 再配布の経過時間を示す. 並列数(MPIプロセス数)の増加に伴い, 固有値計算と再配布の経過時間は局所的に大きく変動する. 固有値計算の最小値は並列数9であり, 固有値計算と再配布を合わせた総経過時間の最小値は並列数256である. 総経過時間を最小化する並列数を設定し, 固有値計算の並列条件を最適化する. 同一次元数の対称密行列の固有値計算は, 行列の成分に関わらず計算量は不変である. 固有値計算の総経過時間を最小化する並列数は, 任意の対称密行列を用いて事前に抽出可能である.
2.2 Optimization of SCF convergence conditions
次にSCF計算の収束条件の最適化について説明する. 従来のSCF計算手順はFigure 2に示すように, 1)電子密度ρ'(r)の初期設定, 2)ρ'(r)の保存, 3) Kohn-Sham方程式を解き, 4,5,6)電子密度ρ'(r)と電子密度の差分Δρ(r), 全エネルギーEnを計算する, 9)Δρ(r)の収束判定を行い, 未収束の場合は10) ρ'(r)を更新して2)からはじめる. Δρ(r)が微小変動となる場合, 収束条件を満たさずSCF計算の繰返し回数が増加する. これは, 電子密度が小さい点において, 電子密度の差分Δρ(r)が大きくなり発生すると考えられる. 提案手法はFigure 2に示す通り, 「7)全エネルギーの変化率dEの計算, 8) dEの収束判定」を追加し, SCF計算の収束条件を最適化する. 以上により, 電子密度の差分Δρ(r)の微小変動によるSCF計算の繰返し回数の増加を抑止し, SCF計算を加速できると考える.
3 Evaluation
計算化学ソフトウェアの計算アルゴリズムの最適化による高速化手法について, 富岳にも採用されたFujitsu A64FX CPU (2.2GHz, 48 cores, 32GiB HBM2)を搭載するSupercomputer (Table 1a)を用いて評価した. 今回, 使用したソフトウェアはオープンソースソフトウェアのCP2K [4]であり, 計算手法としてDFTが利用可能な計算化学ソフトウェアである. CP2KはTable 1bに示す環境を用いてビルドしたMPIとOpenMPのHybrid MPI版実行モジュールを使用した. 高速化手法の性能測定に用いたアプリケーションモデルは, CP2K同梱ベンチマーク等からDFTと混成密度汎関数法(Hybrid DFT)の合計4種類を使用した. 基底関数は平面波ガウス混合基底(GPW)を使用し, 各モデルの計算手法, Hartree-Fock (HF)の割合, 分子種, 分子数はTable 2に示す通り. 並列実行は, 物理1コアに1スレッドを割当てて実行した. 並列条件は「{ノード数}n{MPIプロセス数}p{MPIプロセスのスレッド数}t」と表記する. 例えば, 8ノード, 各ノードは8MPIプロセス, 各MPIプロセスは6スレッドの場合, 各ノードは実48コアで実行したことになり, 8n64p6tと表記する.

Table 1. Evaluation environment
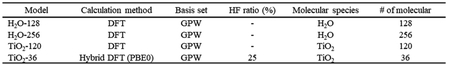
Table 2. Evaluation data of CP2K
Table 3は固有値計算の並列条件の最適化をDFTに適用した場合の並列条件, 総経過時間に対する固有値計算の経過時間の割合, 固有値計算とDFT全体の加速率を示す. 固有値計算の加速率は最大1.48倍, DFT全体の加速率は最大1.33倍, 固有値計算の経過時間の割合が大きい場合DFT全体の加速率は大きくなる. この結果は, DFTにおける固有値計算の並列条件の最適化の有効性を示している.
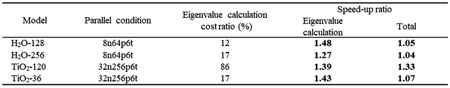
Table 3. SCF calculation speedup ratio by optimization of eigenvalue calculation
Table 4はSCF収束条件の最適化をDFTに適用した場合のSCF計算の繰返し回数, 全エネルギー, 加速率を示す. 並列条件は32n256p6t, dEの収束条件は小数点以下5桁である. SCF計算の繰返し回数は最大約60%減少し,DFT全体は最大2.51倍加速し, 電子密度の差分Δρ(r)が微小変動を繰り返す場合, DFT全体の加速率は大きくなる. また, 全エネルギーは小数点以下6桁以上一致している. この結果は, DFTにおけるSCF計算の収束条件の最適化の有効性を示している.

Table 4. SCF calculation speedup ratio by optimization of convergence conditions (tolerance of dE: 1.0e-5)
4 Conclusion
今回, 固有値計算の並列条件の最適化, SCF計算の収束条件の最適化, これら2つの計算アルゴリズムの最適化による高速化手法を計算化学ソフトウェアCP2KのDFT, Hybrid-DFTに実装して有効性を確認した. CP2Kのアプリケーションモデルを用いて性能測定を実施し, 固有値計算の並列条件の最適化の加速率1.33倍, SCF計算の収束条件の最適化の加速率2.51倍が得られることが示された. また, 材料探索の効率化は, 計算化学ソフトウェアの高速化とAI技術を組合わせた, 探索時間を短縮する新材料探索技術の検討が必要と考え, 今後検討を行う予定である.
References
- [1]
S. Ramakrishna, T.-Y. Zhang, W.-C. Lu, Q. Qian, J. S. C. Low, J. H. R. Yune, et al., J. Intell. Manuf., 30, 2307 (2019). doi:10.1007/s10845-018-1392-0
- [2]
D. S. Sholl, J. A. Steckel, Density functional theory: a practical introduction. John Wiley & Sons, 2011.
- [3]
T. Harville, M. S. Gordon, J. Chem. Theory Comput., 17, 6910 (2021). doi:10.1021/acs.jctc.1c00705 PMID:34699218
- [4]Kühne, Thomas D., et al. "CP2K: An electronic structure and molecular dynamics software package-Quickstep: Efficient and accurate electronic structure calculations." The Journal of Chemical Physics 152.19 (2020): 194103.