Abstract
π-conjugated carbon molecules such as fullerenes and carbon nanotubes have attracted
great interest because of their potential applications as electronic materials and
catalysts. Among these molecular frameworks, fullerene substructures of sumanene molecules
with C3v symmetry are expected to
form vertical columnar crystal structures and exhibit a range of conduction properties.
Various syntheses of molecules with extended π-conjugation at the benzyl position of
sumanene have now been reported. However, there are no examples of open-shell systems with
extended sumanene features. In this study, we use the results of DFT calculations to
discuss the relationship between molecular conformational symmetry and spin multiplicity
in a new, extended open-shell system of sumanenes by adding a phenyl group at the benzyl
position.
1 Introduction
In recent years, organic π-conjugated compounds, the basic materials for practical
applications in organic electronics such as organic transistors and organic EL, have
attracted much attention in the field of novel molecular design and functional expression
[1,2,3,4]. π-conjugated
compounds exhibit optical absorption and luminescence properties due to the conjugation of π
electrons and can conduct electricity by controlling intermolecular interactions and carrier
generation. Since π-conjugated molecules generally have planar structures, new electronic
materials based on molecules with nonplanar structures, such as fullerene C60 are
expected to have unique electronic and physical properties derived from their 3D π-electron
conjugated structures. Among them, the buckybowl-type compound sumanene
(C21H12) [5], which
corresponds to the partial structure of fullerenes, has the following characteristics: (i)
the benzyl position is an sp3-hybridized carbon atom, which makes it easy to
functionalize via radicals and cations; and (ii) in the crystalline state, it forms a
column-like stacked structure derived from the bowl shape, and exhibits conductive
properties [6, 7]. In this regard, sumanene is essential for creating new bowl-shaped
compounds.
The column structure can be controlled by introducing various substituents at the benzyl
position, and a range of extended sumanene derivatives have been reported to date [8]. However, there are no examples of open-shell systems
in extended sumanene derivatives. In this study, we discuss the relationship between
molecular conformational symmetry and spin multiplicity of a new sumanene derivative with a
phenyl group at the benzyl position (triphenyl sumanene; see Figure 1) based on DFT calculations.
2 Method
Calculations were performed with the Gaussian 16. The DFT method was used to optimize the
structure and to calculate the vibrational frequencies of triphenyl sumanene radical
molecules at the B3LYP/6-31G (d,p) level. The B3LYP functional has been used extensively and
has given the most reliable results for structure prediction of organic radical molecules
[7].
3 Results and Discussion
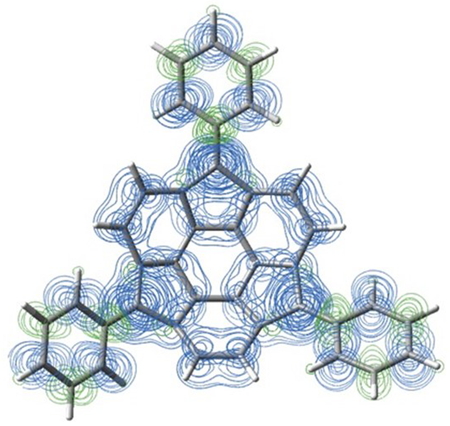
The triphenyl sumanene radical molecule (Figure
1) considered in this study has triply degenerate SOMO localized at the three
equivalent benzyl positions and has a quartet spin multiplicity, judging from the
C3v symmetry structure of the
original sumanene. However, steric hindrance between the phenyl groups causes the phenyl
groups to twist and change orientation, thereby affecting the conformational symmetry and
spin multiplicity of the radical molecule.
The optimized structure for the quartet state is shown in Figure 2. The sumanene structure was curved to ≈ 27°, yielding a
structure with C1 symmetry (Figure 2 (a)), with one phenyl group twisted disrotatory (Tw-Dis), and a structure
with C3 symmetry (Figure 2
(b)), with three phenyl groups twisted conrotatory (Tw-Con). The spin density
distribution of Tw-Con (Figure 3) shows that the
spins localize symmetrically at the three benzyl positions of sumanene. A comparison of the
total energies (Table 1) revealed that Tw-Dis is
slightly more stable by 0.005 eV and that the difference in the orientation of the twist of
the phenyl group is small.
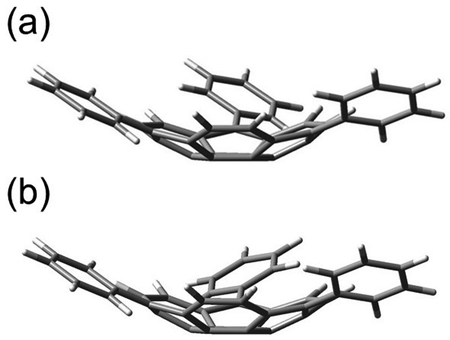
Table 1.
Total Energy, Relative Energy and Relative Energy with zero-point
correction
|
Doublet |
Quartet |
| Total Energy |
Relative Energy |
Relative Energy with ZPC |
Total Energy |
Relative Energy |
Relative Energy with ZPC |
| (Hartree) |
(eV) |
(eV) |
(Hartree) |
(eV) |
(eV) |
| Tw-Dis |
−1498.7592 |
−0.933 |
−0.906 |
−1498.7248 |
0.003 |
−0.005 |
| Tw-Con |
−1498.7577 |
−0.893 |
−0.873 |
−1498.7249 |
0.000 |
0.000 |
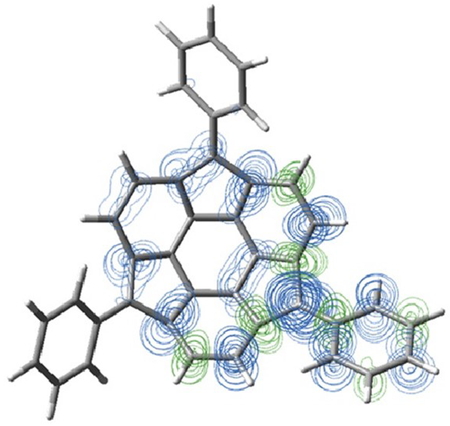
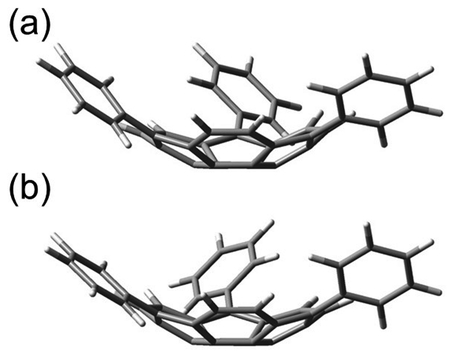
The optimized structure for the doublet state, in which two of the three spins are
singlet-coupled, is shown in Figure 4. As
described for the quartet state, a structure with C1 symmetry,
in which one phenyl group is twisted disrotatory (Tw-Dis, Figure 4 (a)), and a structure in which all phenyl groups are twisted conrotatory
(Tw-Con, Figure 4 (b)), were obtained. The latter
structure had C1 symmetry with slightly broken
C3 symmetry. Both structures are curved by ≈ 30°, reflecting
the increase in the sp3 nature of carbon atoms. Comparing the energies (see Table 1), we find that Tw-Dis is 0.040 eV (0.033eV
with zero-point correction) more stable, as is the case for the quartet state, and that the
difference in the orientation of the twist of the phenyl group has a more significant effect
on the total energy for the doublet. The two unpaired electrons next to each other coupled
antiferromagnetically rather than ferromagnetically in the ground state, resulting in the
doublet energy being significantly lower than the quartet energy. The spin density
distribution of Tw-Con shows (Figure 5) that a
spin is localized in the benzyl position where the twist of the phenyl group is smaller than
the others. This energy stabilization is due to a structural change involving symmetry
breaking, i.e. the Jahn-Teller effect [9].
In summary, the relationship between the symmetry of the molecular structure and the spin
multiplicity of triphenyl sumanene radical molecule as a novel sumanene derivative radical
molecule is discussed based on DFT calculations. It is found that the effect of the phenyl
group on the total energy is more significant for the doublet state compared with the
quartet state. It may be expected that it is easier to create a stable stacked structure for
quartet multiplicity since pairs of stacking radical molecules can easily adjust their
structures. We plan to study the structure symmetry and spin multiplicity of stacked systems
of triphenyl sumanene radical molecules in the future.
Acknowledgment
We would like to thank Prof. K. Yamashita for fruitful discussions from experimental and
theoretical perspectives. This work was supported by JSPS KAKENHI Grant Number 20A201. The
computations were performed at the Research Center for Computational Science, Okazaki, Japan
(Project: 22-IMS-C064) and the Center for Computational Materials Science, Institute for
Materials Research, Tohoku University for the use of MASAMUNE-IMR (MAterials science
Supercomputing system for Advanced MUlti-scale simulations toward NExt-generation-Institute
for Materials Research).
References
- [1] H. Sakurai, T. Daiko, T.
Hirao, Science, 301, 1878 (2003). doi:10.1126/science.1088290
- [2] A. Kasprzak, A. Tobolska, H.
Sakurai, W. Wróblewski, Dalton Trans., 51, 468 (2022).
doi:10.1039/D1DT03467G
- [3] Q. Tan, P. Kaewmati, S.
Higashibayashi, M. Kawano, Y. Yakiyama, H. Sakurai, Bull. Chem. Soc. Jpn., 91, 531 (2018).
doi:10.1246/bcsj.20170384
- [4] A. Muraoka, M. Hayashi,
Chem. Phys. Lett., 748, 137393 (2020). doi:10.1016/j.cplett.2020.137393
- [5] M. Saito, H. Shinokubo, H.
Sakurai, Mater. Chem. Front., 2, 635 (2018). doi:10.1039/C7QM00593H
- [6] T. Amaya, S. Seki, T.
Moriuchi, K. Nakamoto, T. Nakata, H. Sakane, et al., J. Am. Chem. Soc., 131, 408 (2009).
doi:10.1021/ja805997v
- [7] T. Amaya, T. Hirao, Chem.
Commun. (Camb.), 47, 10524 (2011). doi:10.1039/c1cc12532j PMID:21743888
- [8] T. Amaya, H. Sakane, T.
Muneishi, T. Hirao, Chem. Commun. (Camb.), 2008, 765 (2008).
doi:10.1039/B712839H
- [9] K. Raghavachari, R. C.
Haddon, T. A. Miller, V. E. Bondybey, J. Chem. Phys., 79, 1387 (1983).
doi:10.1063/1.445897