Abstract
接着剤分子を新たに設計する際には,
理論と実験の両面からその接着機構に関して考察することが重要である.本研究では,新規接着剤分子である2-メルカプトピリジル基(2MP基)をもつビニルモノマーについて,被着体表面との吸着状態を解析することを最終目的とした.同モノマーに対し密度汎関数法に基づいた第一原理量子力学計算を実行し,その分子軌道を算出した.2MP基の平衡反応(互変異性化反応)によって得られるチオール型とチオン型の電子状態を比較したところ,HOMOとLUMOの分子軌道の状態から,被着体表面と分子軌道を介して相互作用を起こすときはチオン型の方が優位であることが確認された.
Translated Abstract
In recent years, adhesives have been studied and used in many fields. Both basic and
applied researches have been conducted. But, the adhesion mechanism is still not
understood in detail. The purpose of this study is to analyze the adsorption state of a
vinyl monomer having a 2-mercaptopyridyl (2MP) group. The vinyl monomer is used as a
component of adhesive materials. 2MP groups attach to the surface of the adherend, and
they undergo tautomerization reactions where thione- or thiol-type chemical structures are
taken. Computational analyses were performed according to density general function method
and the first principles of quantum mechanics.
We calculated the molecular orbitals of vinyl monomers of including the
2MP group. Our evaluation revealed the electronic states of each chemical structure
through the equilibrium reaction.The influence of the electronic states on the adsorption
by 2MP groups was then analyzed and compared.
1 諸言
現代においては,接着剤が様々な分野で使用され, 基礎・応用両面の研究が進められている [1].私たちの身の回りにある接着剤は, 各種の原子や分子によって構成されており, それらと接着対象(被着体)表面との相互作用によって様々な吸着状態が生じている.しかし,
その接着機構に関しては不明な点も多い.接着剤分子と被着体表面に生じる吸着状態を決定づけているのはそれら分子の電子状態であると考えられるが,
接着機構の解明には実験事実を積み重ねるだけでなく, ミクロな視点での考察が必要となってくる.そのための知見を得る有効な手段の一つとして,
コンピュータ上に対象となる分子を再現するコンピュータシミュレーションがある [2,3,4,5,6].コンピュータシミュレーションの中で,固体や液体などの凝縮系の電子状態を算出する際,モデル化せずに多体問題にも対応して扱おう手法として密度汎関数理論(Density
Functional Theory, 以降 DFT)を用いた第一原理計算が有効ある [7,8,9].
日本におけるDFT計算の例としては, 佐藤文俊らによるタンパク質の全電子シミュレーションや宇田毅, 加藤弘一らのシリコン表面酸化の様子のシミュレーションなどがあり,
その他にも様々な分子や系に対してシミュレーションが行われ, 議論されている [10, 11].また,接着剤分子としてはエポキシ樹脂が広く用いられており,エポキシ樹脂の接着に関するDFT計算としては,辻雄太らからエポキシ樹脂とグラファイトに関する接着,尾形修司らからエポキシ樹脂のアミン基のプロトン化について報告されている.
[12, 13]
本研究では,エポキシ樹脂に代わる新たな接着分子として期待される2-メルカプトピリジル(2MP)基を持つビニルモノマーを研究対象とした.この接着分子はラジカルUV硬化系を用いる光接着材料であり,ガラスや金属に対して強い接着力を有することが報告されている
[14].この接着力ついては,2MP基が異なる被着体表面において平衡反応により分子構造を変化させることで,吸着状態を最適化させることに起因するのではないかと実験結果から示唆されているが,接着の微視的メカニズムを実験的調査によって明らかにすることは困難である.そこで,平衡反応(互変異性化反応)に伴って生じるチオン型及びチオール型の分子構造(Figure 1 (a)
チオン型,(b)チオール型)に対して分子軌道計算を行い,算出したそれぞれの電荷の状態及び分子軌道から,被着体表面と接着層の界面付近において2MP基のどちらの構造が優位となるかということについて明らかにすることを目的とした.なお,この接着材料では対象分子の他に2-ヒドロキシエチルメタクリレート(HEMA,
Figure 2)が併用されているので,HEMAに対しても分子軌道計算を行った.
2 計算手法
本研究は,Windows10上でWinmoster V10.7.0 (株式会社クロスアビリティより購入) [15]を用いてGAMESSを実行した.
Winmosterは, 量子化学計算, 分子道力学計算, 固体物理計算をマウス操作で実行するためのオールラウンド型シミュレーションソフトウェアである.
GAMESS (General Atomic and Molecular Electronic Structure Sys-tem) [16, 17]は,
Gaussianと並び広く利用されている非経験的分子軌道法計算パッケージである.GAMESSでは, 閉殻/開殻系のハートリー・フォック法(HF), 密度汎関数法(DFT),
一般化原子価結合法(GVB), 多配置SCF法 (MCSCF)を含む一般的な量子化学計算を実行することができ,
双極子モーメントから周波数依存超分極率までの広い範囲にわたる様々な分子物性を求めることができる.GAMESSには多くの基底関数が内蔵されており,
さらに外部から基底関数を読み込むことも可能なため, 本質的には周期表のすべての原子に対する計算が可能である.電子相関補正は, 配置間相互作用(CI),
Moller-Plesset摂動法(MPn)などにより見積もることができ, さらに溶媒効果や相対論的補正を含めた計算も可能である [18].
3 計算手順
接着対象分子を解析するために行った計算の一連の流れを示す.モデリングから電荷計算, 被着面の形成までの方法や条件設定の方法などを説明する.
3.1 モデリング
Figure 1 (a)のチオン型分子の分子モデルを作成した(Figure 3).
3.2 最適化・軌道計算
モデリングした分子を用いて最適化を行った.
最適化において最も重要なのは初期構造である.実験値がない場合, 計算負荷が軽い低レベルの方法であらかじめ最適化を行い, 初期構造を作る.非常に大きい分子系の場合,
全体の最適化を最初に行うより, 末端を水素原子などで適当に置き換えた部分構造を先に最適化し, それを初期構造に使うという方法が有効である.これにより,計算負荷の軽減や収束が良くなる
[18].よって, 本研究でも, Figure 3の窒素原子を炭素原子に置き換え, 酸素原子や硫黄原子なしの状態でSTO-3G計算を繰り返し,
全体の最適化構造に至った.その後, HF/6-31Gおよび HF/6-31G*で軌道計算を行った.Figure
4にチオン型対象分子の最適化構造と各原子の電荷の大きさを表示したもの(赤:負電荷,青:正電荷)を示す.
同様に, チオール型対象分子のモデリング, 構造最適化計算によって得られた分子モデルと,軌道計算によって得られた各原子の電荷の大きさを表示したものをFigure 5に示す.
4 結果
本研究で得られた電荷(Mulliken電荷)と分子軌道 (HOMO (Highest Occupied Molecular Orbital)とLUMO (Lowest
Unoccupied Molecular Orbital))の計算結果について,以下に示す.
4.1 全エネルギー
チオール型およびチオン型対象分子の構造最適化計算から算出された全エネルギーは,STO-3G軌道計算ではチオール型が-1143.526 kcal/molでチオン型-1143.508
kcal/molであった.HF/6-31G軌道計算では,チオール型が-1156.955 kcal/mol,チオン型が-1156.960
kcal/mol.HF/6-31G*軌道計算では,チオール型が-1157.304 kcal/molで,チオン型が-1157.281 kcal/molであった.
4.2 電荷
まず,チオン型対象分子のMulliken電荷についてFigure 6に示す.
次に, チオール型対象分子のMulliken電荷をFigure 7に示す.
4.3 分子軌道
本研究での計算では, 電荷の他に分子軌道のそれぞれの相対的なエネルギーとその時の分子軌道の形を得ることができた.
まず,チオン型対象分子について得られた軌道データをTable
1に,チオール型対象分子について得られた軌道データをTable
2にそれぞれ示す.
Table 1.
Molecular orbitals and relative energies of thione-type target molecule
| Item |
Result |
| HOMO |
No.71 |
| LUMO |
No.72 |
| HOMO-LUMO Gap |
8.6723 [eV] |
| HOMO Energy |
−7.2736 [eV] |
| LUMO Energy |
1.3987 [eV] |
Table 2.
Molecular orbitals and relative energies of thiol-type target molecule
| Item |
Result |
| HOMO |
No.71 |
| LUMO |
No.72 |
| HOMO-LUMO Gap |
10.8437 [eV] |
| HOMO Energy |
−8.3131 [eV] |
| LUMO Energy |
2.5307 [eV] |
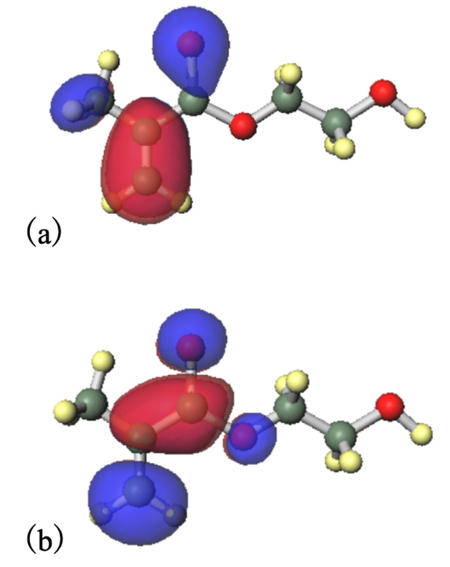
また, 得られた分子軌道の形をFigure 8に示す.
5 HEMA
併用されるHEMA分子は接着材料中,HEMA:2MP = 100:1.3
(モル比)の割合で配合されており,接着材料の主成分と言える.よって,接着機構解明においてHEMA分子に関しても調べることは必須であるため,分子軌道計算を行った.
Figure 9にHEMAの最適化された分子モデルを示す.
そして, HEMAについて得られた分子軌道データをTable 3に,分子軌道の形をFigure 10にそれぞれ示す.
Table 3.
Molecular orbitals and relative energies of HEMA
| Item |
Result |
| HOMO |
No.35 |
| LUMO |
No.36 |
| HOMO-LUMO Gap |
11.7689 [eV] |
| HOMO Energy |
−10.4601 [eV] |
| LUMO Energy |
1.3089 [eV] |
6 考察
得られた結果から,電荷と分子軌道について順に考察する.
6.1 全エネルギー
STO-3G軌道計算,HF/6-31G軌道計算,HF/6-31G*軌道計算それぞれの全エネルギーの算出結果結果から,チオール型とチオン型のエネルギー差は最大でも0.026
kcal/molであり,全エネルギーに対して0.002%程度の極僅かな違いであり,チオール型およびチオン型において構造的に優位な構造はないことが明らかとなった.このことから,対象分子の平衡状態は周囲の環境に適した構造へ移動するものと思われる.
また,チオール型およびチオン型の全エネルギーについてSTO-3G軌道計算とHF/6-31G軌道計算結果を比較すると,HF/6-31G軌道計算結果がチオール型は13.42
kcal/mol,チオン型は13.45
kcal/molだけ安定化する結果となった.さらに,HF/6-31G軌道計算結果とHF/6-31G*軌道計算結果を比較すると,HF/6-31G*軌道計算結果がチオール型は0.345
kcal/mol,チオン型は0.320 kcal/molだけ安定化していた.
STO-3G軌道計算とHF/6-31G軌道計算から,対象分子の分子軌道の広がりに自由度を持たせた方が精度が向上し,更にHF/6-31G軌道計算とHF/6-31G*軌道計算から,電荷の偏りなどにおいては分極関数を加えなくともHF/6-31G軌道計算である程度説明できる.よって,本研究の目的の精度と計算コストを考慮し,HF/6-31Gを用いることとした.
6.2 電荷
STO-3G軌道計算,HF/6-31G軌道計算,HF/6-31G*軌道計算それぞれの電荷の算出結果を比較したところ,Mulliken電荷の値に大きな変化は確認されなかった.また,Lowdin電荷についても算出すると,Lowdin電荷ではMulliken電荷に比べて電荷の値が全体的に小さくなる傾向が見受けられたが,極性が変化するようなことないことが確認された.
Figure
4右図から,チオン型の対象分子ではアミド結合部の窒素原子に大きな負の電荷を,炭素原子に大きな正の電荷をそれぞれ有していることが確認された.また,2MP基部分に着目すると,窒素原子に-0.783
eの電荷が確認され,この窒素原子に結合する水素は0.455 eの電荷がそれぞれ確認された.さらに,チオカルボニル部分に着目しても,硫黄原子に-0.314 e,炭素原子に0.229
eの電荷がそれぞれ確認された.
一方,Figure
5右図から,チオール型の対象分子でもチオン型と同様にアミド結合部に最大の電荷の偏りが確認された.アミド結合部の窒素原子が大きな負電荷,炭素原子が大きな正電荷をそれぞれ有していることがわかった.2MP基部分では,窒素原子に-0.578
eの電荷が確認されたものの,チオン型対象分子よりはやや小さいことがわかった.さらに,チオール部分に着目すると,硫黄原子(0.0790 e),硫黄原子に隣接する炭素原子(0.121
e),水素原子(0.109 e)であり,いずれもほぼ無電荷であることがわかった.
以上のことから,対象分子のアミド結合部は互変異性化反応に依らず,常に極性官能基として働くことが確認された.また,対象分子の2MP基部分の窒素,炭素,硫黄,水素の各原子における電荷の状態より,チオール型よりもチオン型の方が電荷の偏りが顕著であるため,ガラスのように表面が強く分極した被着体表面と相互作用を起こしやすいと考えられる.これは,X線光電子分光法(XPS)により接着層破断面の分子状態を調べた実験結果と,矛盾しない考察となる
[14].
6.3 分子軌道
HOMOについては,Figure 8 (a)
Thione-HOMOから,チオン型では2MP基部分にその分子軌道が確認された.一方,チオール型では,Figure 8 (c)
Thiol-HOMOから,ビニル基部分にその分子軌道が確認された.このことから,対象分子の2MP部分が電子リッチな官能基として被着体表面に供与作用するのはチオン型のときだけであり,チオール型では起こり得ないことが明らかになった.
LUMOについては,Figure 8 (b)
Thione-LUMOから,チオン型では2MP基部分に分子軌道が確認された.一方,チオール型においても,Figure 8 (d)
Thiol-LUMOから,2MP基部分に分子軌道が確認された.さらに,ビニル基にも分子軌道が確認された.よって,互変異性化反応後の分子の形によらず,2MP基部分が被着体表面から電子を受け取るような形での相互作用が起こり得ることが明らかとなった
[19,20,21,22].
以上のことから,表面が強く分極した被着体表面と2MP基部分の間で相互作用するときには,チオン型が優位であることが示唆された.
次に,HEMA分子の分子軌道計算結果(Figure
10)より,HEMA分子が重合後に高分子側鎖となる部分にはいずれの分子軌道も確認されなかった.このことからHEMA由来側鎖の分子軌道が被着体表面との相互作用を起こす可能性は低いと思われる.しかし,HEMA分子のビニル基には,HOMO・LUMOとも大きな分子軌道が確認されている.したがって,HEMA分子同士や同じくビニル基に分子軌道が確認されるチオール型の対象分子とビニル結合をおこし,鎖状分子の形成に大きく寄与していると考えられる.
7 結言
本研究では,光接着材料の構成要素である2MP基を持つビニルモノマーを研究対象とし,この対象分子の平衡反応(互変異性化反応)により生じるチオン型及びチオール型に着目した.これらに対してそれぞれ分子軌道計算を行い,算出したそれぞれの分子軌道から,被着体表面と接着層の界面付近において対象分子が取る優位な構造はどちらかということについて明らかにした.さらに,接着材料中で併用されるHEMAに対しても分子軌道計算を行った.
電荷については,チオン型,チオール型ともアミド結合部分に最大の電荷が確認され,これは平衡反応に対して不変であった.チオン型では,2MP基部分の窒素原子に負の電荷,水素原子に正の電荷を有していることが確認され,チオール型でも,2MP基部分の窒素原子に負の電荷が確認された.しかし,チオン型の窒素原子の負電荷はチオール型よりも大きく,水素原子の正電荷の影響もあり,チオン型の方が被着体表面と静電的な相互作用を起こしやすいと考えられる.
分子軌道については,チオン型のみHOMOで2MP基部分に分子軌道が見られた.また,LUMOについてはチオン型,チオール型共に2MP基部分で分子軌道が見られた.このことから,被着体表面と分子軌道を介して相互作用を起こすときはチオン型の方が優位であることが確認された.
本研究によって2MP基を持つビニルモノマーの接着には軌道相互作用と静電的な相互作用が重要であることが明らかとなった.しかし,接着には水素結合やOH-π相互作用,溶媒分子を含めた影響なども重要であることが提唱されており
[23],これら要因を考慮した計算を実行することが今後の課題である.
参考文献
- [1] The Adhesion Society of Japan,
ed., Sechakuhandbook Dai 4 han. Nikkan Kogyo Shinbun (2007).
- [2] R. Maezono, T. Ichiba,
Ugokasite rikaisuru daiichigennridennshijotaikeisan, Morikita Publishing Co.
(2020)
- [3] A. Ueda, Konpyutashimyureshon
-Makuro na kei no naka no gennsiundou -, Asakura Publishing Co. (1991).
- [4] I. Okada, ed., Eiji Oosawa, ed.,
Bunshi shimyureshon nyuumon, Kaibundo Co. (1989).
- [5] L. Schlangen, F. A. M.
Leermakers, L. K. Koopal, J. Chem. Soc., Faraday Trans., 92, 579 (1996).
- [6] R. Car, et al., Phys. Rev.
Lett., 5, 2417 (1985).
- [7] M. Ruike, Fundamentals of
Quantum Chemistry, Tokyo Denki University Press (2013).
- [8] D. W. BALL, Trans., K.Tanaka, T
Atake, Ball buturikagaku jyou, Kagaku-Dojin Publishing Co. (2015).
- [9] R. G. Parr, W. Yang, Trans.,
Satoru Kano, et al., Density-Functional Theory of Atoms and Molecules, Springer Japan
(1996).
- [10] F. Sato, J. Comput. Chem.,
6, 145 (2007).
- [11] T. Uda, Koichi Kato, Oyo
Buturi, 66, 1199 (1997).
- [12] Y. Tsuji et al., ACS
omega 4.3, 4491 (2019).
- [13] S. Ogata et al., The
Journal of Physical Chemistry B 125.31, 8989 (2021).
- [14] M. Furutani, D. Fujihira, K.
Arimitsu, J. Photopolym. Sci. Technol., 33, 261 (2020).
- [15] R. Koga, CICSJ Bull., 33,
109 (2015).
- [16] M. J. Perri, S. H. Weber, J.
Chem. Educ., 91, 2206 (2014).
- [17] M. W. Schmidt, K. K.
Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, et al., J. Comput.
Chem., 14, 1347 (1993).
- [18] K. Hirao, An Easy Guide to
Quantum Chemistry Calculations, Kodansha Ltd. (2006).
- [19] Y. Kunisada, Low
temperature science, 78, 181 (2020).
- [20] K. Masuda, et al. KOBE
STEEL, Eng. Rep., 51, 40 (2001).
- [21] Y. Kunisada, J. Vac. Soc.
Jpn., 60, 249 (2017).
- [22] J. Neugebauer, M. Scheffler,
Phys. Rev. B Condens. Matter, 46, 16067 (1992).
- [23] S. Nakamura, Y. Tsuji, K.
Yoshizawa, ACS Omega, 5, 26211 (2020).