S0/S1極小エネルギー円錐交差(MECI)の支配因子を評価するために,スピン反転時間依存密度汎関数理論に対する凍結軌道解析(FZOA)の波動関数と励起エネルギーを導出した.スピン反転法に特有のスピン汚染を避けるため,定式化においてスピン完全法を適用した.数値計算の結果,「HOMO−LUMO交換積分がほぼ0となる」,「HOMO−LUMOギャップの上限値はHOMO,LUMOが関係するCoulomb積分によって定まる」というS0/S1 MECIの支配因子を発見した.本論文では,FZOAについて概説するとともに,スピン反転法におけるFZOAの定式化について述べる.導出したスピン反転FZOAの式をエチレンとウラシルに適用した結果に基づいて,励起エネルギー成分に基づく支配因子の発見,S0/S1 MECIの電子構造,これらの制限付き開殻法における結合係数への依存性について解説する.
We have derived the wavefunction and excitation energy of frozen orbital analysis (FZOA) for spin-flip time-dependent density functional theory in order to evaluate the controlling factor of the S0/S1 minimum energy conical intersection (MECI). The spin-complete method was applied in the derivation to avoid the spin contamination inherent in spin-flip approaches. From the results of numerical calculations, the controlling factors of S0/S1 MECI were found as "HOMO−LUMO exchange integral is close to zero" and "Coulomb integrals related to HOMO and LUMO determine the upper limit of the HOMO−LUMO gap". In this paper, we account for the FZOA and its formulation within the spin-fip method. Based on the application of the derived formula of spin-flip FZOA to ethylene and uracil, we explain the discovery of the controlling factors based on the excitation energy components, the electronic structure of the S0/S1 MECIs, and their dependence on the coupling coefficients of the restricted open-shell method.
円錐交差(CI)は光励起した分子の失活過程において重要であり,その分子構造は平衡構造や遷移状態と大きく異なる.一般的に有機分子は基底状態で一重項であり,多くの場合,光励起後は速やかにS1状態に遷移することから,S0状態とS1状態間の極小エネルギーCI (S0/S1 MECI)が特に重要である.中井らは,凍結軌道解析(FZOA) [1,2,3,4,5,6] を時間依存密度汎関数理論(TDDFT)に基づく量子化学計算に対して行った.その結果,有機分子のS0/S1 MECI構造において「最高占有分子軌道(HOMO)と最低非占有分子軌道(LUMO)の交換積分がほぼ0となる」,「HOMO−LUMOギャップとHOMO−LUMO Coulomb積分がほぼ等しい値になる」という支配因子を発見した [7, 8].
これらの研究は閉殻基底状態を参照状態とするTDDFTを対象としているが,MECIの構造最適化計算では,MECI近傍のポテンシャルエネルギー面を記述可能であるスピン反転TDDFT (SF-TDDFT) [9, 10]が用いられる.SF-TDDFTではS0/S1 MECIにおいて交換積分が0に近い傾向は確認されたが,HOMO−LUMOギャップとHOMO−LUMO Coulomb積分がほぼ等しい傾向は見られなかった [11].我々は,SF-TDDFTに対してFZOAを行い支配因子の再評価を行った.その結果,「HOMO−LUMO交換積分がほぼ0となる」という先行研究 [7, 8]と同じ支配因子に加えて,「HOMO−LUMOギャップの上限値はHOMO,LUMOが関係するCoulomb積分によって定まる」という支配因子を発見した [12].
本論文では,SF-TDDFTにおけるMECI構造の支配因子の発見について解説する.初めに支配因子を見つける手がかりとして用いたFZOAについて解説し,SF-TDDFTに対するFZOAの定式化を説明する.スピン反転法に基づくFZOAによる励起エネルギーの式をエチレンとウラシルに適用した場合の数値的結果についても触れる.スピン反転法によらない場合と数値を比較することで,TDDFTとSF-TDDFTで異なる支配因子が見出された理由を考察する.原著論文 [12]では触れられていないFZOAにより得られる電子状態についても考察する.
FZOAは,最小の活性空間で波動関数を表現することで電子状態間のエネルギー差を簡潔かつ解釈が容易な式で表し,この式を数値的に評価することで電子励起について考察する解析法である.分子軌道(Kohn−Sham DFTではKohn−Sham軌道)は状態ごとに最適化することはせず,参照状態の量子化学計算で決定した軌道をそのまま用いる.
簡単な例として,Hartree−Fock計算による閉殻基底状態を参照状態としてHOMOからLUMOへの1電子励起配置で記述される一重項励起状態を考える.HOMOとLUMOがどちらも縮重していない場合,最小活性空間はHOMOとLUMOで構成される2電子2軌道の空間となる.Figure 1に示す2つの配置状態関数よりハミルトニアン行列HFZOAを構築すると
| (1) |
となる.ここでEは基底状態のエネルギー,ΔεHLはHOMO−LUMOギャップである.JHLはHOMO−LUMO Coulomb積分,KHLはHOMO−LUMO交換積分であり,それぞれ
| (2) |
| (3) |
と表される.ここで,φHとφLはそれぞれHOMOとLUMOの空間軌道である.JHLはHOMOの電子とLUMOの電子との古典的Coulomb相互作用のエネルギーであり,KHLはHOMOの電子とLUMOの電子との量子力学的相互作用を反映する.ハミルトニアン行列の非対角項はBrillouinの定理より0となる.式(1)より基底状態から一重項励起状態への励起エネルギー
| (4) |
となる.式(1)は活性空間を制限しない配置間相互作用法におけるハミルトニアン行列の一部である.ゆえにFZOAを配置間相互作用法の近似として捉えることができる.

The singlet configuration state function of FZOA for the closed-shell reference wavefunction.
この考えに基づいてDFTの枠組みにおけるFZOAをTDDFTの近似として定式化できる.TDDFTによる励起エネルギーωはCasidaの式で与えられる.Tamm−Dancoff近似のもとでは
| (5) |
となる.ここでAは軌道Hessian行列と呼ばれる行列であり,Xは各電子配置の寄与を表すベクトルである.参照状態をDFT計算で得られる閉殻基底状態とし,A行列のうちFigure 1に示した配置状態関数(Referenceを含む)のみ考慮すると,FZOAによる励起エネルギーωFZOAが
| (6) |
| (7) |
と導出される.cHFは交換相関汎関数におけるHartree−Fock交換項の割合であり,EXCは交換相関汎関数におけるHartree−Fock交換項以外の部分,ρは電子密度,φはKohn−Sham軌道である.式(6)においてcHF = 1,EXC = 0とおくと励起エネルギーは式(4)に帰着する.
式(4)と式(6)はそれぞれHartree−Fock法,Kohn−Sham DFTによる参照状態に適用される式であるが,両者はHOMO−LUMOギャップの値が大きく異なることがよく知られている.系の電子数がNであるとき,Hartree−Fock法では,占有軌道の電子はその電子以外のN−1電子によるポテンシャルの影響を受けるが,仮想軌道はN電子によるポテンシャルにより決定される.一方,Hartree−Fock交換を混成しないKohn−Sham DFTでは,原理的には占有軌道,仮想軌道はどちらもN−1電子によるポテンシャルにより決定される.この差異は仮想軌道のエネルギーに影響する.また,電子相関がHartree−Fock法では含まれずDFTでは含まれることと,実際のDFT計算では交換相関汎関数の近似により自己相互作用が生じることにより占有軌道のエネルギーにも違いが生じる.以上の要因によりHartree−Fock法によるHOMO−LUMOギャップは一般的にKohn−Sham DFTより顕著に大きい.近似交換相関汎関数におけるHartree−Fock交換項の混成比率は,軌道エネルギーやHOMO−LUMOギャップの値に大きく影響する.
FZOAによる励起エネルギーに関しては,交換相関汎関数としてB3LYPなどの一般的な汎関数を用いると式(7)の右辺第2項は0に近い値となり,HOMO−LUMO Coulomb積分はcHFでスケーリングされるため,式(6)のJ'HLは式(4)のJHLより小さくなる.ゆえにFZOAによる励起エネルギーの電子状態理論や汎関数への依存性は,軌道エネルギーの依存性よりは小さい.この依存性については,文献 [5]にてCO分子の計算例が示されている.この例では電子励起に関与する軌道が縮重しているため,FZOAによる励起エネルギーの式は式(4)および式(6)とは異なることに注意されたい.
文献 [7]では式(6)に基づき種々の有機分子に対してωFZOA,ΔεHL,J'HL,KHLが評価された.BHHLYP汎関数を用いた計算の結果,S0/S1 MECIにおいて以下の2つの傾向が見出され,閉殻基底状態を参照状態とするTDDFTにおけるS0/S1 MECIの支配因子として報告された.
| (8) |
| (9) |
これらの支配因子は数値計算により経験的に見出されたものであるが,MECIにおいて式(8)が成立し,なおかつS1状態がFigure 1の|1Φ>でよく近似できるのであれば,式(6)にてωFZOAとKHLがほぼ0となり式(9)が成立することがわかる.
以上をふまえた上でSF-TDDFTにおけるFZOAを考える.SF-TDDFTでは参照状態として高スピン状態のDFT計算を行い,1電子をスピン反転励起させた電子配置により基底状態や励起状態を記述する.これにより閉殻電子配置からの2電子励起配置が取り込まれ,二重結合の回転や円錐交差近傍のポテンシャルエネルギー面が定性的に正しく記述される.Tamm−Dancoff近似のもとで,SF-TDDFTによる参照状態からの励起エネルギーωは式(5)で与えられる.式(5)におけるA行列は
| (10) |
となる.ここでFはKohn−Sham軌道を基底とするFock行列の成分であり,↑と↓はそれぞれアップスピン,ダウンスピンを表す.本論文では,制限付き開殻法で得られる三重項波動関数を参照波動関数とし,参照波動関数を構成する半占有軌道を便宜上HOMO,LUMOと呼称する.
SF-TDDFTに対するFZOAとしてHOMO,LUMOのみを活性空間とすると,軌道Hessian行列はFigure 2の電子配置を基底とする4×4の行列となる.これを対角化すると4つの電子状態が得られるが,式(4)や式(6)のように状態間のエネルギー差をHOMO−LUMOギャップや積分にあらわに依存した式として簡潔に表すことができない.また,得られる4つの状態はスピン二乗演算子の固有関数とはならい.すなわちスピン汚染が生じるため,電子状態の解釈が困難となる場合がある.
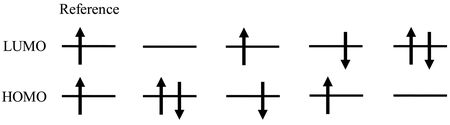
Four electronic configurations within the minimum active space of SF-TDDFT as well as the reference state.
そこで,スピン完全(SC)法 [13]に基づきスピン反転励起による一重項配置状態関数(式(11)−(13))を基底とした軌道Hessian行列ASC-SF-FZOAを構築する.
| (11) |
| (12) |
| (13) |
とはそれぞれ生成演算子,消滅演算子である.これ以降,スピン反転法とSC法に基づくFZOAをSC-SF-FZOAと略記する.これらの電子配置はFigure 3により容易に理解できる.
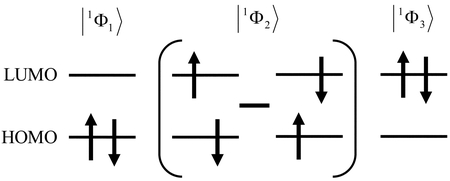
Singlet configuration state functions of SC-SF-FZOA.
一重項と三重項のカップリング項を無視するため,ASC-SF-FZOAの対角化によりスピン汚染のない一重項状態が得られる.
制限付き開殻法で参照波動関数を得る場合,FZOAによる励起エネルギーを軌道エネルギー差に依存した式で表すために,軌道エネルギーの任意性に対処する必要がある.三重項参照状態のFock行列成分とそのHOMOエネルギーεHおよびLUMOエネルギーεLの間には以下の関係がある.
| (14) |
| (15) |
AOO = BOO ≠ 0のとき,ASC-SF-FZOAにおけるFock行列成分を軌道エネルギーを用いた表式に置き換えることができる.Roothaan [14]もしくはGuest−Saunders [15]による結合係数(AOO = BOO = 1/2)を採用すると,
| (16) |
となる.ここでΔεHL = εH − εLであり,J"とK"はそれぞれHOMO,LUMOに関するCoulomb積分,交換積分の寄与であり,以下の式で与えられる.
| (17) |
| (18) |
ΔεHLおよびK"は0以上,J"は0以下 [16]の値をそれぞれとる.HOMOとLUMOの空間的な重なりが小さいほどJ"は負に大きくなり,K"は0に近づく.
スピン反転FZOAによるS0,S1,S2状態の波動関数は式(16)の固有ベクトルより,以下の通りとなる.
| (19) |
| (20) |
| (21) |
固有値の差より,スピン反転FZOAによるS0-S1およびS0-S2エネルギー差の式はそれぞれ式(22),(23)となる.
| (22) |
| (23) |
本論文では,実際のエネルギーの順番によらず式(19),式(20)をそれぞれS0,S1状態として扱う.S0状態がS1状態より高いエネルギーとなる場合は,式(22)で表される状態間のエネルギー差を負数で表す.
なお,McWeeny−Diercksenによる係数 [17]もAOO = BOOを満たし,これを用いると式(16)および以降の式におけるΔεHLに係数3/2が掛けられる.それぞれの結合係数をTable 1にまとめた.
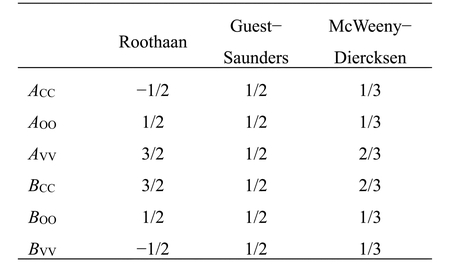
このようにSF-TDDFTに対してもFZOAによる励起エネルギーをHOMO−LUMOギャップとCoulomb積分,交換積分で表すことができる.閉殻基底状態を参照状態とする場合,HOMOとLUMOの占有数をそれぞれ2,0として軌道を最適化する.一方,制限付き開殻法では軌道の最適化においてHOMOとLUMOの占有数を1とする.軌道エネルギーの結合係数への任意性に加えて参照状態の計算における占有数の違いにより,HOMO−LUMOギャップの数値は交換相関汎関数や基底関数を揃えても全く違う値になりうることには注意が必要である.
エチレンとウラシルについて,S0状態の平衡構造(S0 EQ)をBHHLYP/6-31G(d,p),その他の構造をSF-TD-BHHLYP/6-31G(d,p)による構造最適化計算で得た.SC法に基づくスピン反転FZOA (以降,単にFZOAと呼ぶ)により,励起エネルギーとその成分を計算した.交換相関汎関数はBHHLYP (cHF = 0.5),基底関数は6-31G (d,p)を用いた.計算はGAMESSプログラム [18]を用いて行った.
初めにエチレンについて,Roothaanの結合係数を用いて計算を行った.Table 2にSF-TDDFTとFZOAによるS0-S1励起エネルギーと,FZOAの式に現れるエネルギー成分について,S0 EQ,炭素-炭素結合を90°回転させたねじれ構造(Twisted),S0/S1 MECIに対してそれぞれ計算した結果を示す.各構造におけるKohn−Sham軌道をFigure 4に示す.S0 EQではHOMO−LUMOギャップや交換積分が大きいため,S0-S1エネルギー差は大きい.ねじれ構造ではHOMOとLUMOが縮退する.HOMO−LUMOギャップの減少は状態間エネルギー差の減少に大きく寄与するが,交換積分が大きいためS0状態とS1状態は縮退せず,エネルギーギャップは大きい.

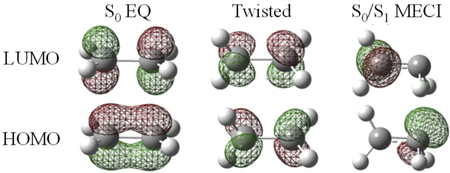
Kohn−Sham orbitals of ethylene.
MECIでは対称性の低下によりHOMOとLUMOの分布は重なりが小さくなる.HOMOは炭素原子の2p軌道,LUMOはsp3混成軌道と同様の分布である.Coulomb積分の寄与が負に大きく,交換積分の寄与はほぼゼロとなる.対称性の低下による軌道の局在化を伴う変化(sudden polarization) [19]によりHOMOとLUMOの空間上の重なりが小さくなることで,Coulomb積分や交換積分の励起エネルギーへの寄与が減少し,状態間のエネルギー差が小さくなることがわかる.
Table 3には,FZOAによるS0状態について電子配置の係数を示した.式(19)より,係数はHOMO−LUMOギャップと交換積分から決定される.S0 EQでは,S0状態は|1Φ1>の寄与が支配的であるが,2電子励起配置の係数もある程度の値をとる.これは,S0状態は共有結合性であるが,イオン性をわずかに含むことを意味する(文献 [20]のFigure 3を参照).S1状態は|1Φ2>で与えられるので,Figure 3とFigure 4よりS1状態はπ-π*励起状態である.ねじれ構造ではHOMOとLUMOが縮退しているので,2つの電子配置の寄与は等しい.この構造では,S0状態はジラジカル状態,S1状態は双性イオン状態と解釈される [20].S0 EQでは2電子励起配置の寄与があるが,MECIではその寄与はほとんどない.すなわち,MECIにおいてS0状態が近似的に|1Φ1>で表される.この電子配置はHOMOの占有数が2,LUMOの占有数が0でありFigure 4に示した軌道との対応により,S0状態は双性イオン状態と解釈される.また,|1Φ2>はHOMO,LUMOどちらも占有数が1であり,軌道との対応からS1状態はジラジカル状態と解釈される.
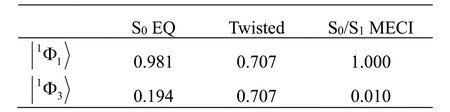
続いてウラシルについて3種類の結合係数を用いた計算を行い,S0-S1励起エネルギーの成分とS0状態の波動関数における係数をTable 4とTable 5にまとめた.RoothaanとGuest−Saundersの結果はほぼ一致した.McWeeny−Diercksenの結合係数を用いるとHOMO−LUMOギャップは他の結合係数の場合より小さい値となるが,HOMO− LUMOギャップに依存する量であるFZOAのS0-S1エネルギー差や波動関数における電子配置の係数は,他の結合係数の結果とよく一致した.


S0 EQ (Table 4)ではHOMO−LUMOギャップや交換積分が大きく,Coulomb積分の寄与は小さい.S0/S1 MECI (Table 5)では,交換積分がほぼ0となり,軌道エネルギー差とCoulomb積分の寄与の和が負となるため,S0-S1エネルギー差は負となる.交換積分やCoulomb積分がS0 EQとS0/S1 MECIで大きく異なることは,MECIにてHOMOとLUMOの空間的な重なりが小さいこと(Figure 5)と対応している.これらの傾向はエチレンのS0 EQとS0/S1 MECIと同様である.
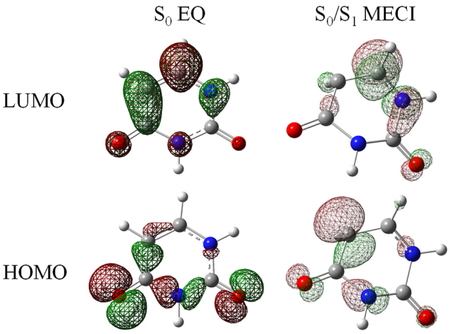
Kohn−Sham orbitals of uracil. Carbon, hydrogen, nitrogen, and oxygen atoms are shown in gray, white, blue, and red, respectively.
波動関数の係数とKohn−Sham軌道の分布より,S0 EQにおけるS0状態からS1状態への電子励起はn-π*励起と解釈される.S0/S1 MECIでは,交換積分が小さいことからS0状態はほぼ単一の電子配置で記述される.HOMOとLUMOの空間的な重なりが小さいため,S0状態はHOMOに2つの電子が入った双性イオン状態,S1状態はHOMOとLUMOに電子が1つずつ入ったジラジカル状態として解釈される.以上より,SF-TDDFTによるウラシルのS0/S1 MECIは,エチレンの場合と同様に双性イオン状態とジラジカル状態のエネルギーが縮退する構造と理解された.
閉殻基底状態を参照状態とするFZOA (以降,単にFZOAと呼ぶ)では,S0/S1 MECIにおいてHOMO−LUMO交換積分がほぼ0となり,式(6)の右辺はΔεHL − J'HLとなる.開殻三重項状態を参照状態とする場合(SC-SF-FZOA)では,Table 2およびTable 5よりS0/S1 MECIにおいてHOMO−LUMO交換積分がほぼ0となり,式(22)の右辺はRoothaanもしくはGuest−Saundersによる結合係数を用いた場合,ΔεHL + J"HLとなる.
ここで,S0/S1 MECIにおけるHOMO−LUMOギャップ(ΔεHL)とCoulomb積分の寄与(−J'HL,J"HL)を比較する.Figure 6は文献 [7, 12]にて報告された31種類,42個のMECIをBHHLYP汎関数と6-31G (d,p)基底関数系を用いて計算した結果である.Figure 6よりFZOAではHOMO−LUMOギャップとCoulomb積分の寄与が相殺することがわかる.これは,HOMO−LUMO交換積分に関する支配因子に加えて式(6)の左辺が0に近いためである.FZOAではFigure 1の電子配置のみ考慮しており,この取り扱いはMECIではTDDFTに対するよい近似となり,S1状態のエネルギーをほとんど変化させないため,式(6)の左辺が0に近くなる.
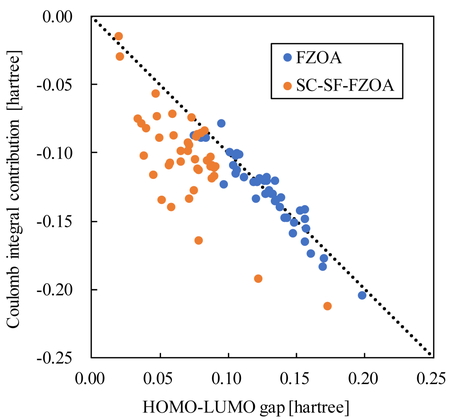
一方,SC-SF-FZOAではHOMO−LUMOギャップとCoulomb積分の寄与が絶対値で同程度の場合もあれば,Coulomb積分の寄与がより負に大きい場合もある.FZOAによる活性空間の制限およびスピン対称性を満たす波動関数の取り扱いがMECIにおけるS0状態とS1状態のエネルギーを同じ値だけシフトさせる場合,式(22)の左辺におけるHOMO−LUMOギャップとCoulomb積分の寄与が釣り合う.このシフトがS0状態の方が正に大きい場合,式(22)の左辺は負となり,HOMO−LUMOギャップとCoulomb積分の寄与の和が負となる.結果としてSC-SF-FZOAではS0/S1 MECIの支配因子として式(8)に加えて
| (24) |
が見出された.式(24)は,MECIにおけるHOMO−LUMOギャップの上限値はHOMO,LUMOが関係するCoulomb積分によって定まることを意味している.
なお,Figure 6よりSC-SF-FZOAではFZOAと比較してHOMO−LUMOギャップが小さい傾向が確認でき,参照状態の計算方法の違いが大きく異なる軌道エネルギーを与えることを示している.HOMO−LUMOギャップの違いにより,FZOAにおける軌道エネルギーに関する支配因子はSC-SF-FZOAでは成立しない.
本論文で説明したMECI構造における支配因子は数値計算により帰納的に見出されている.これまでの研究では有機分子のS0/S1 MECIに注目し計算を行ってきたが,MECIに限らずS0状態とS1状態が縮退する空間の広範囲において同じ関係が成立するかなど,さらなる検証が求められる.一方,我々は交換積分がほぼ0になるという支配因子には何らかの理由が存在し,支配因子を理論的に導出できる可能性があると考えている.軌道エネルギーに関する支配因子は交換積分が0になるという支配因子と,FZOAによる波動関数の取り扱いがMECIにおける状態間のエネルギー差に影響する程度によって成立すると考えられる.
本論文では,SF-TDDFTに対するFZOAに基づく支配因子の発見について解説した.初めに閉殻基底状態を参照状態とするFZOAについて概説し,SF-TDDFTに対するFZOAの定式化を説明した.制限付き三重項波動関数を参照状態とし,SC法に基づき波動関数が一重項スピン対称性を満たすようにした.さらに軌道エネルギーの結合係数が特定の条件を満たす場合,状態間のエネルギー差がHOMO−LUMOギャップおよびHOMO,LUMOに関する積分を用いて簡潔に表された.
導出された式をエチレンとウラシルに適用した場合の数値的結果も示した.S0/S1 MECIにおいて「HOMO−LUMO交換積分が0に近い(
本研究で得られた成果は,SF-TDDFT計算による光物性や光化学反応の解析に有用と考えられる.さらに,我々はMECIの構造最適化計算において支配因子を拘束条件に加えることを報告しており [11],支配因子の発見が量子化学計算の方法論の発展に寄与することも期待している.
本研究は,科学研究費基盤研究(C)(研究課題番号21K05002)の支援を受けて実施された.本研究で行った量子化学計算の一部は,自然科学研究機構(NINS)計算科学研究センター(RCCS)の計算機を利用した(Project: 23-IMS-C047).