Abstract
Octanol/water partition coefficient, log P, is an important parameter in classical QSAR. The new method using machine learning which we propose uses only the molecular orbital energy as an explanatory variable and does not include log P. Therefore, since the log P value can be predicted using the molecular orbital energy, we speculated that log P may not be necessary as a result if sufficient number of molecular orbital energies would be given as parameters.
Translated Abstract
Octanol/water partition coefficient, log P, is an important parameter in classical QSAR. The new method using machine learning which we propose uses only the molecular orbital energy as an explanatory variable and does not include log P. Therefore, since the log P value can be predicted using the molecular orbital energy, we speculated that log P may not be necessary as a result if sufficient number of molecular orbital energies would be given as parameters.
1 はじめに
近年ニューラルネットの発展である深層学習により,機械学習が再び注目を集め化学分野への応用の可能性も広がってきている.我々は分子軌道法により求められた分子軌道エネルギーが説明変数として使えるのではないかと考え,一連の研究を行ってきた [1,2,3,4,5].
我々は,2008年にベンゾジアゼピンおよびチエノジアゼピン系の抗不安薬について,抗不安性と抗痙攣性の強さとnext-HOMOの軌道エネルギー値と相関があることを見いだしたことから,このような発想に至った [1].以前の発表でフェルラ酸(FA)の抗酸化作用に対して,DPPHフリーラジカル消去濃度(IC50)の置換基効果を説明できることを示した [2, 3].ただし,ただ一つの軌道エネルギー値に対して強い相関関係を得ることは一般的には難しいため,複数の軌道エネルギー値とIC50との関係をRandom Forest回帰法による機械学習を試みたところ,強い相関関係が見いだされ,フェルラ酸の抗酸化作用に関する構造活性相関については分子軌道計算のみでIC50値の予測が可能となった.さらに,ブースティングやニューラルネットなどRandom Forest以外の回帰法を用いて機械学習を行った結果についても報告している [4, 5].
一方,このような薬理活性相関の研究においては,Hansch-FujitaによるQSAR法が以前より使用されてきた.QSAR法では,リーガンド・レセプター間で様々な分子間相互作用を考え,それぞれを表す記述子を使用することにより構造活性相関が見積もられる.記述子としてはHOMOおよびLUMOのエネルギー値も含まれているが,非常に重要なパラメータとして,脂溶性を示すオクタノール/水分配係数,log Pがある.
我々が提案している機械学習は分子軌道エネルギーだけを説明変数としておりlog Pは含まれていない.そこでlog P値は分子軌道エネルギーを用いて予測できるので結果としてlog Pを必要としないのではないかと推測して今回検討を行ったので報告する.
2 計算方法
分子軌道計算プログラムGaussian16 RevB.01を使用しRHF/6-31G(d,p)レベルで構造最適化を行った [6].最適化された構造に対して振動数計算を行い,安定構造であることを確認した.
機械学習計算にはRのCaretパッケージ [7]を使用し分子軌道計算により得られた軌道エネルギー値を説明変数として,最大80軌道までを使用して解析を試みた.トレーニングデータは16分子,未知データとして扱うテストデータは6分子とした.データの分割は,乱数を使用して行った.Validationは3-fold-validationを使用した.回帰法は,HYFIS, SBC, WM, gamboost, glm, kknn, krlsRadial, lasso, monmlp, pls, ppr, qrf, ranger, rf, svmLinear, svmRadial, xgbLinear, xgbTreeの18種類を比較した.
トレーニングデータとテストデータを合わせたものに対する決定係数Xが最も大きくなる時の分子軌道エネルギーの数を説明変数の最適な数とした.テストデータの予測値と実験値の差のroot mean square (RMS値)が最も小さくなる時の軌道エネルギーの数も参考とした.なおXは相関係数の自乗と等しい.
対象とする分子とlog Pの値はRekkerらの論文 [8]にあげられている実験値により定められた22種類を用いた.Atropine, Chloramphenicol, Chlorothiazide, Chlorpromazine, Cimetidine, Diazepam, Diltiazem, Diphenhydramine, Disopyramide, Flufrenemic acid, Furosemide, Haloperidol, Imipramine, Lidocaine, Phenobarbital, Phenytoin, Procainamide, Propafenone, Propranolol, Tetracaine, Trimethoprim, Verapamilである.ただしこれらのうち,Chlorothiazide, Chlorpromazine, Diazepam, Disopyramide, Furosemide, Haloperidol, Propafenone, Propranololの8種類についてはPubChem [9]に収録されているものと異なるため,PubChemの値を採用した.
3 結果と考察
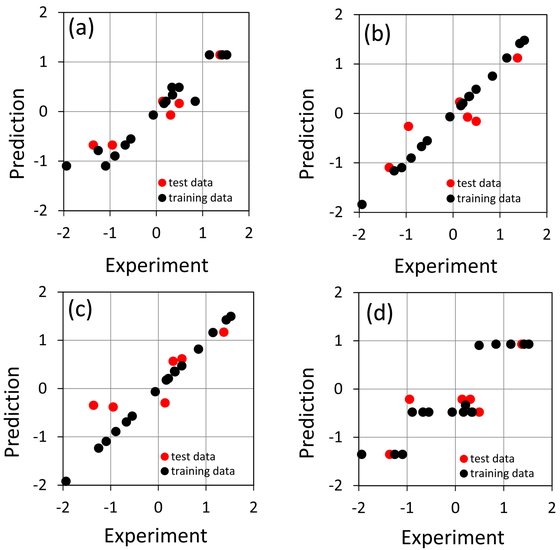
Table. 1に2~80個の分子軌道数で,テストデータの決定係数X,テストデータも含めた場合の決定係数X,トレーニングデータの決定係数X,テストデータの予測値と実験値との誤差のRMSEの最小値を18種類の回帰法について示した.またFigure1には実験値と予測値の分散を示した.予測値,実験値,RMSEの各数値は標準化されている.
Table 1. X values of test, total, and training data, and RMSE of test data
| Method | Number of Orbitals | X test | X total | X training | RMSE |
| qrf | 2 | 0.904 | 0.919 | 0.927 | 0.378 |
| xgbLinear | 4 | 0.793 | 0.943 | 0.999 | 0.445 |
| xgbTree | 10 | 0.778 | 0.922 | 1.000 | 0.527 |
| WM | 10 | 0.671 | 0.764 | 0.797 | 0.588 |
| rf | 2 | 0.619 | 0.796 | 0.867 | 0.624 |
| HYFIS | 14 | 0.698 | 0.791 | 0.893 | 0.674 |
| ranger | 8 | 0.554 | 0.806 | 0.922 | 0.691 |
| SBC | 16 | 0.530 | 0.861 | 0.999 | 0.699 |
| gamboost | 4 | 0.446 | 0.659 | 0.767 | 0.732 |
| lasso | 58 | 0.489 | 0.807 | 0.959 | 0.735 |
| pls | 68 | 0.385 | 0.710 | 0.831 | 0.745 |
| svmRadial | 16 | 0.464 | 0.732 | 0.857 | 0.760 |
| krlsRadial | 24 | 0.373 | 0.827 | 1.000 | 0.772 |
| kknn | 10 | 0.382 | 0.616 | 0.723 | 0.798 |
| svmLinear | 60 | 0.378 | 0.776 | 0.992 | 0.892 |
| monmlp | 64 | 0.242 | 0.705 | 1.000 | 1.104 |
| ppr | 62 | 0.503 | 0.132 | 1.000 | 2.313 |
| glm | 36 | 0.637 | 0.385 | 1.000 | 4.211 |
テストデータのXの値はHOMO/LUMOの2軌道のエネルギーを説明変数としqrfを回帰法とした場合の0.904が最大となっていてその時のRMSEは0.378で最小となっている.xgbLinearを回帰法として軌道数4の場合に0.793になっている.この時のRMSエラー値は0.445となっている.その次はxgbTreeの0788であるが,これは軌道数が10の時の値である.RMSエラー値は0.527とやや大きくなっている.4番目はWMの0.671であるが軌道数がxgbTreeと共に10と大きくなっている.WMの予測結果はほぼ3種類の予測値だけとなっていて興味深い.今後の検討が必要であろう.その他の結果から概ね,boosting系とrandom forest系の回帰法が少ない軌道数で良い結果を与えているように思われる.
一方,線形重回帰であるglmはXの最大値は0.637と一見良さそうに見えるがtraining dataに対するXが1となっていることからもわかるように過学習の結果であり,テストデータのRMSEが4.221となっていて全く予測ができていないことがわかる.部分的最小二乗回帰plsではXの最大値が0.385で弱い相関関係しか得られなかった.また軌道数も68で非現実的な値となっている.
4 結論
本研究では,機械学習を用いてQSAR法で使用されるオクタノール/水分配係数を目的変数とし分子軌道のエネルギー値を説明変数として予測する事を試みた.
18種類の回帰法を使用し,また説明変数の数を従来の方法ではほぼ不可能であった80種類まで増加させて計算することにより,多くの回帰法と説明変数を与えることで最適な結果が得られることがわかったが,その結果,回帰法としてqrfを使用しHOMO/LUMOの軌道エネルギーだけを説明変数とするだけで最適な解を得ることができた.その他の回帰法としては勾配boostingならびにrandom forestが有効であり,線形回帰や部分線形回帰では満足な結果は得られなかった.
参考文献
- [1]
H. Teramae, K. Ohtawara, T. Ishimoto, U. Nagashima, Bull. Chem. Soc. Jpn., 81, 1094 (2008). doi:10.1246/bcsj.81.1094
- [2]
H. Teramae, M. Xuan, T. Yamashita, J. Takayama, M. Okazaki, T. Sakamoto, J. Comput. Chem. Jpn., 17, 150 (2018). doi:10.2477/jccj.2018-0018
- [3]
H. Teramae, M. Xuan, T. Yamashita, J. Takayama, M. Okazaki, T. Sakamoto, J. Comput. Chem. Jpn., 18, 211 (2019). doi:10.2477/jccj.2019-0034
- [4]
H. Teramae, T. Matsuo, K. Niwatsukino, R. Inoue, S. Noguchi, M. Xuan, et al., J. Comput. Chem. Jpn., 19, 43 (2020). doi:10.2477/jccj.2020-0005
- [5]
H. Teramae, M. Xuan, J. Takayama, M. Okazaki,T. Sakamoto, AIP Conf. Proc., 2611, 020007 (2022). doi:10.1063/5.0119589
- [6]Gaussian 16, Revision B.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- [7]
https://cran.r-project.org/web/packages/caret/caret.pdf
- [8]
R. F. Rekker, A. M. T. Laak, R. Mannhold, Quant. Struct.-Act. Relationsh., 12, 152 (1993). doi:10.1002/qsar.19930120207
- [9]
PubChem, https://pubchem.ncbi.nlm.nih.gov/