速報 (SCCJ Annual Meeting 2023 Spring Poster Award Article)
量子化学計算と各種スペクトル情報を用いた化合物の自動同定手法の開発
2023 年 22 巻 2 号 p. 12-14
詳細
2023 年 22 巻 2 号 p. 12-14
Recent practical application of automated experiments using robotics, high-throughput experiments, and artificial intelligence technology has been progressing rapidly. In automated experiments, molecular identification is an important process for obtaining structural information on synthesized compounds and understanding their reactivity and chemical properties. In this study, we developed a system for automated molecular identification. The system uses spectral information and quantum chemical calculations, which provide no fluctuating data and have a potential to explore a wide range of chemical space. Numerical validation results suggested that the system is capable of efficient and accurate automated molecular identification in organic compounds with low molecular weight.
Recent practical application of automated experiments using robotics, high-throughput experiments, and artificial intelligence technology has been progressing rapidly. In automated experiments, molecular identification is an important process for obtaining structural information on synthesized compounds and understanding their reactivity and chemical properties. In this study, we developed a system for automated molecular identification. The system uses spectral information and quantum chemical calculations, which provide no fluctuating data and have a potential to explore a wide range of chemical space. Numerical validation results suggested that the system is capable of efficient and accurate automated molecular identification in organic compounds with low molecular weight.
近年,ロボティクス技術やハイスループット実験,人工知能(AI)技術を活用した自動実験の実用化が急速に進展している.自動実験のサイクルにおいて,化合物同定は合成された化合物の構造情報の取得や,反応性・物性などを理解するための重要な工程である.従来の化合物同定は,人間が核磁気共鳴(NMR)スペクトルなどの各種スペクトルから得た構造情報と矛盾しないように,化合物を予測してきた.このような人間による化合物同定の正確さや速さは,一般的に研究者の経験と勘に大きく依存する.
この工程をAIが代替する場合,膨大な化学空間からスペクトルなどの情報を用いて,如何に一つの化合物に絞り込むかが問題となる.1968年以来,自動的に化合物同定を行うための様々な手法が開発されてきた [1,2,3,4].これらの手法は,入力に用いるスペクトル情報の種類,同定対象(低分子有機化合物,タンパク質など),データベースの利用の有無,ルールベースや機械学習のシステムの違いなどにより分類される [5].近年においても,化学空間全体から効率的に探索し,かつ高精度に自動同定できるシステムを目指した研究が進められている.
既存の自動的な化合物同定手法の多くは,実験データを利用している.しかし,実験データは実験者や実験条件の違いに起因する揺らぎを含む.そのため,機械学習などの予測精度を低下させる可能性がある.そこで本研究では,データの揺らぎがない量子化学計算を活用して,自動的に化合物同定を行う手法を開発した.また量子化学計算は既知・未知問わず任意の化合物を考慮できるため,広範の探索を可能にする.
Figure 1に提案するシステムの流れを示す.まず,①スペクトル情報から部分構造を抽出する.次に,②部分構造同士を組み合わせて化合物を生成し,分子量が質量スペクトルの結果と一致する化合物を候補として残す.③候補となる化合物すべての安定構造を探索し,④量子化学計算により算出した化学シフト値を参照スペクトルの値と比較する.最後に,⑤値の一致度に基づいて候補化合物をランク付けし,同定結果を出力する.

A schematic flow of proposed system.
本研究では特に,スペクトルの測定誤差や分析の影響を排除し,②~⑤の流れの妥当性を検証するため,以下のような代替を行った.①の部分構造は,IRスペクトルなどから抽出する代わりに,化合物データベースPubChemからSMILES表記で化合物構造を取得し,BRICS分解 [6]して得た.②の質量スペクトルに対応する情報は,PubChemから分子量を取得した.NMRスペクトルの参照値は,量子化学計算によるデータベースであるPubChemQC [7]からωB97X-D/6-311G (d,p)レベルで構造最適化された三次元座標データを取得し,密度汎関数理論(DFT)により核磁気遮蔽定数を算出することで得た.
本システムの各過程における具体的な手法は以下の通りである.②の候補となる化合物を生成するためにBRICSBuild [6]を使用した.③の安定構造探索では,autodE [8]により自動生成された複数の配座から,構造最適化後のエネルギーが最も低い配座を安定構造として使用した.構造最適化計算には,DFTまたはニューラルネットワークポテンシャルのPreferred potential (PFP) [9]を使用した.PFP計算はMatlantis version 3.0.0 [10]のMOLECULEモードで実行した.④の核磁気遮蔽定数の比較では,DFTによる計算値と参照値の間の平均二乗偏差(RMSD)を指標とした.DFT計算はすべてB3LYP/6-31G(d,p)レベルで行った.なお将来的に実験値と比較することを考慮し,構造最適化は揺らぎを含んだ構造と比較するために参照と異なるレベルで計算した.一方,核磁気遮蔽定数は構造が同じならば,ほぼ同じ値を与えると仮定し,参照と同じ計算レベルで行った.
本研究では,PubChemQCに含まれる化合物のうち,H,C,N,O,F,S,Cl,Brの8原子からなる化合物をランダムに40個抽出し,本システムに適用した.すべての化合物に対する結果の詳細をSupplementary Materials (SM)にまとめる.その結果,30個の化合物は②の候補化合物の生成段階において一つの化合物のみとなった.これらはすべて目的となる化合物と一致した.
残りの10個の化合物のうち,2個の化合物(ID1とID2)の結果をFigure 2に示す.括弧外はDFT,括弧内はPFPのRMSDである.ID1とID2はそれぞれ候補化合物が6個であった.橙色の点線は目的となる化合物を示す.この結果,ID1,ID2ともにDFTとPFPの両方で目的となる化合物が一位となり,正しく同定できることが確認された.また,SMに詳細を示すように,その他の8個の化合物についても正しく同定できた.RMSDがゼロにならないのは,参照における構造最適化の計算レベルが異なることに起因しており,構造がある程度一致していれば,化合物同定ができることを示している.

Results of present compound identification in ranking format for (a) ID1 and (b) ID2. The RMSD values in DFT (bracket-free) and PFP (in parentheses) are shown.
続いて,本システムにおける計算時間について議論する.Figure 3にID1の各ステップにおける計算時間の割合を示す.なお構造最適化はDFTの結果である.この結果,構造最適化計算が全体の72.3%を占めた.この構造最適化における,DFTとPFPの計算時間をFigure 4に示す.この結果から,PFPはDFTよりも約56倍高速であり,より効率的な化合物同定が可能であることが示唆された.
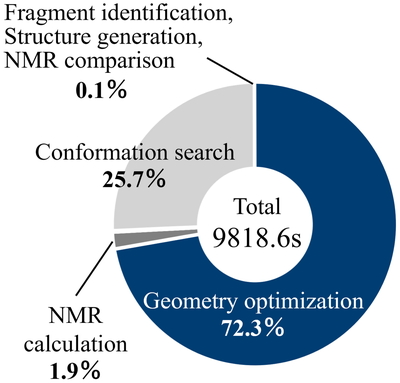
Percentage of CPU time for each step in ID1.
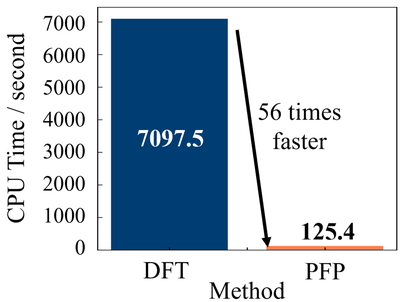
CPU times for geometry optimizations with DFT and PFP in ID1.
本研究では,スペクトルまたはそれと類似の情報と,量子化学計算を組み合わせた化合物の自動同定システムを開発した.量子化学計算を活用することで,候補化合物から対象となる化合物の構造を正確に予測できた.また,DFTの代わりにPFPを用いることで,化合物同定の精度を落とすことなく,効率的な構造最適化が実現された.本システムは,既知未知問わず効率的かつ高い精度で化合物を自動的に同定できる可能性があることが示唆された.
この研究は独立行政法人日本学術振興会(JSPS)科学研究費(JP21K04998) の補助を受けている.また,量子化学計算の一部は計算科学研究センター(RCCS)の計算機を利用して行った.