Abstract
The molecular descriptor set suggested in electronic-structure informatics (ESI) was
applied to predict activity of potential candidates of α-glucosidase inhibitors. In this
study, we constructed a regression model for predicting the pIC50 values of the
known inhibitors registered in the ChEMBL database. The obtained regression model
reasonably reproduced the experimental values. After refinement of the model, we conducted
in silico screening to search for active inhibitors drugs from a
natural product database, called KampoDB, for traditional Japanese medicine. We have
discovered some promising compounds as potential α-glucosidase inhibitors.
Translated Abstract
The molecular descriptor set suggested in electronic-structure informatics (ESI) was
applied to predict activity of potential candidates of α-glucosidase inhibitors. In this
study, we constructed a regression model for predicting the pIC50 values of the
known inhibitors registered in the ChEMBL database. The obtained regression model
reasonably reproduced the experimental values. After refinement of the model, we conducted
in silico screening to search for active inhibitors drugs from a
natural product database, called KampoDB, for traditional Japanese medicine. We have
discovered some promising compounds as potential α-glucosidase inhibitors.
1 はじめに
α-glucosidaseは糖の吸収や排泄を調節する働きを持つ加水分解酵素である.この酵素の機能を阻害することによって血糖値の急激な上昇が抑えられるため,α-glucosidase阻害剤の探索は糖尿病治療薬の開発における重要な研究課題である.この種の阻害剤のいくつかは既に上市されているが,発展途上国において増加する罹患者数への懸念や,副作用の問題から,現在でもより安全な新規阻害剤の探索が盛んに行われている
[1].
本研究では,天然物の中から新規α-glucosidase阻害剤を探索する目的で,電子状態インフォマティクス(ESI) [2]に基づく機械学習とin
silicoスクリーニングを行った.天然物に注目する理由は,過去に承認された糖尿病治療薬の92%(58種)が天然物または天然物に由来することにある [3].一方,天然物からなる化学空間のin
silicoスクリーニングでは,従来にはない母骨格の探索 ("Scaffold Hopping" [4])が期待される.このため,本研究では,電子状態的特徴を数値化するESIに基づく阻害剤探索を行った.
2 計算方法
2.1 データ収集
ChEMBL データベース [5]から605個のα-glucosidase阻害剤のSMILES表示およびその活性値(IC50)を得た.検索は,"target_chembl_id:CHEMBL
3833502 AND standard_type:("IC50")"
をクエリとし,2022年7月1日に実施した.得られた分子は全てα-glucosidaseに結合するものと考えられる.in
silicoスクリーニング用の天然物は漢方薬(生薬)のデータベースであるKampoDB [6]から収集した.
2.2 ESI記述子の計算
我々がこれまで提案してきたESI記述子には次の諸量が含まれる.(1)スピン1重項の電子基底状態(S0状態)での分子体積,(2)分子内包直方体の3辺の長さ,(3)双極子モーメント,(4)水を溶媒とする溶媒和エネルギー,(5)赤外スペクトルの類似度,(6)振動状態の状態密度の類似度,(7)紫外可視吸収スペクトルの類似度,(8)電子励起状態の状態密度の類似度,(9)分子軌道の状態密度(単位エネルギーあたりの分子軌道の数)の類似度,(10)イオン化エネルギー,(11)イオン化後の構造緩和エネルギー,(12)
イオン化状態の最適化構造での脱イオン化後の構造緩和エネルギー,(13)アニオン化エネルギー,(14)アニオン化後の構造緩和エネルギー,(15)
アニオン化状態の最適化構造での脱アニオン化後の構造緩和エネルギー,(16)最低スピン三重項状態(T1)への垂直遷移エネルギー,(17)
T1状態への遷移後の構造緩和エネルギー,(18)
T1状態での最適化構造での脱励起後の構造緩和エネルギー,(19)分子量,の計19種(21個)の記述子である.分子量以外は,電子状態に依存する分子間相互作用のポテンシャル面の形状,電子状態の変化や応答,電子状態変化に伴う構造的な応答,分子の環境変化に伴うエネルギー利得,を数値的に表現するものである.
本研究では,従来用いてきたESI記述子に加えて,(20)スピン1重項励起状態への励起エネルギーΔEn
(n = 1-30)の逆数を記述子に加えた.これは,
分子物性の記述に必要な1次の摂動波動関数が次式で表されることから,予測精度と解釈性の向上が期待されると考えて導入された.
|
ψ =
ψ
0
(
0
)
+
∑
n≠0
⟨
ψ
n
(
0
)
|
X
^
|
ψ
0
(
0
)
⟩
Δ
E
n
ψ
n
(
0
)
|
上述の記述子(分子量を除く)はDFT (M06-2X)/6-31G (d,p)法あるいはTDDFT (M06-2X)/6-31G
(d,p)法による電子状態計算によって数値的に評価した.これらの計算ではGaussian16プログラム [7]を用いた.
S0状態の初期構造は,SMILES表示から得られた三次元構造をMMFF94分子力場によって構造最適化することによって得た.この構造生成と分子力学計算はRDKit
[8]
によって行った.スペクトル類似度は,最も高いpIC50値を与える分子を参照分子として評価された.
2.3 活性予測とin silicoスクリーニングのための機械学習手法
阻害活性の予測のため,全てのデータを標準化したのちに訓練用データセット(訓練セット)とテスト用データセット(テストセット)に分割し,前者を用いたXGBoost回帰 [9]を行った.分割の際は,乱数を使って10000通りの分割を行い,二つのデータセットの統計分布の差が最も小さい分割を採用した.ハイパーパラメータ最適化には5-fold
cross validationを用いた.その後,全てのデータを用いてin silico スクリーニング用の回帰モデルを構築した.
3 結果と考察
3.1 回帰モデルの構築
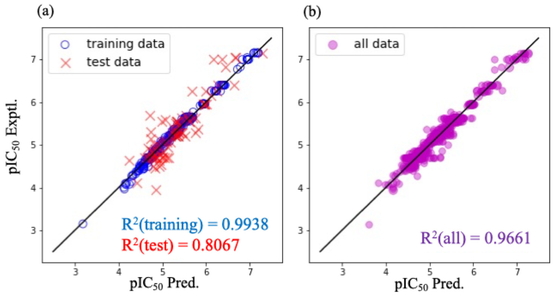
ChEMBLデータベースから得たα-glucosidase阻害剤605分子のうち,計算が完了した433分子のESI記述子を計算し,活性値(pIC50)を予測するモデルを作成した(Figure 1a,Table
1参照).この機械学習では,346分子を訓練セット,87分子をテストセットに含めた.テストセットに対する決定係数(R2)が0.81であることから,回帰モデルによるpIC50の予測値は概ね妥当であると考えられる.
Table 1. Comparison of accuracy of the regression modeling using the ESI, ECFP4, and
RDKit descriptors.
|
ESI desc.
|
ECFP4
|
RDKit desc.
|
|
R2(train)
|
0.9938 |
0.9893 |
0.9933 |
|
R2(test)
|
0.8067 |
0.8607 |
0.8439 |
比較のため,ケモインフォマティクス研究でよく用いられる2種類の記述子(ECFP4とRDKit記述子)による回帰モデルを作成した(Table 1).これらの記述子はいずれもRDKitによって容易に計算することができる.Table
1の結果から,テスト用データに対するR2は,ECFP4によるモデルが最も大きく,次いでRDKit記述子が良好な予測を与えることがわかった.ESI記述子によるモデルの予測性能はこれらによるものと比べやや低いが,0.8以上の値であることから,ESI記述子でも十分な予測性能を有すると考えられる.また,ECFP4やRDKit記述子とは全く異なるESI記述子がそれらと同程度の精度で阻害活性を予測できていることは興味深い.
3.2 天然物に対するin silico スクリーニング
Figure 1aおよびTable 1の結果からESI記述子を用いたXGBoost回帰には汎化性能があると判断し,in
silicoスクリーニングを行った.ここでは,Figure
1aのモデルにテストセットを追加で学習させ,全データにより予測モデルを構築した.このモデルの予測値と実験値の比較から(Figure 1b),その妥当性を確認することができた.
得られたモデルを用いて,KampoDBに登録されている3002分子のうち,電子状態計算が完了した255分子の
pIC50を予測し,有望なα-glucosidase阻害剤を探索した.その結果,Figure 2aに示すpunicalagin,1,2,3,6-tetragalloylglucose などが高活性であることがわかった.Liuら [10],およびBellesiaら [11]によれば,punicalaginは実際にα-glucosidase阻害能を持つと報告されている.一方,Zhangら [12]はミズナラの外皮から抽出された1,2,3,6-tetragalloylglucoseを含むtetragalloylglucoseが
α-glucosidase阻害能を有することを報告している.1,2,3,6-tetragalloylglucoseの阻害能に関する直截的な記載はないが,本研究はそれが活性化合物の候補であることを示唆している.
Figure 2aからわかるように,これらの二分子の構造は,教師データ(Figure
2b)に含まれる高活性分子のそれとは大きく異なっている.これは,ESIによる分子スクリーニングによって "Scaffold Hopping"
が可能であることを示している.
本稿ではESI記述子によるスクリーニングによる結果を示したが,他の記述子によるスクリーニング結果との比較も興味深い課題である.これについては別の機会に報告する.
4 結論
本研究では,α-glucosidase阻害剤の探索を目的として,ESI記述子を用いた XGBoost回帰によるpIC50の予測とin
silicoスクリーニングを行った.実験値と予測値の相関を示す決定係数R2は0.81と比較的高く,良好なモデルを構築できた.スクリーニング用の回帰モデルを構築して生薬データベースから活性分子を探索したところ,教師データの分子とは異なるscaffoldを有する分子が同定された.これはESI記述子を用いた機械学習によって従来にない化合物スクリーニングが可能であることを意味する.
5 謝辞
手法に関する議論をしていただいた大川和史氏,中村登志氏,有意義なコメントをいただいた審査員に感謝いたします.
参考文献
- [1] A. Singh, K. Singh, A.
Sharma, K. Kaur, K. Kaur, R. Chadha, et al., J. Mol. Struct., 1281, 135115 (2023).
doi:10.1016/j.molstruc.2023.135115
- [2] T. Ideo, K. Yoshida, M.
Sugimoto, Chem. Lett., 50, 849 (2021). doi:10.1246/cl.200966
- [3] D. J. Newman, G. M. Cragg,
J. Nat. Prod., 83, 770 (2020). doi:10.1021/acs.jnatprod.9b01285
PMID:32162523
- [4] H. J. Böhm, A. Flohr, M.
Stahl, Drug Discov. Today. Technol., 1, 217 (2004). doi:10.1016/j.ddtec.2004.10.009
PMID:24981488
- [5] D. Mendez, A. Gaulton, A. P.
Bento, J. Chambers, M. De Veij, E. Félix, et al., Nucleic Acids Res., 47, D1, D930 (2019).
doi:10.1093/nar/gky1075 PMID:30398643
- [6] R. Sawada, M. Iwata, M.
Umezaki, Y. Usui, T. Kobayashi, T. Kubono, et al., Sci. Rep., 8, 11216 (2018).
doi:10.1038/s41598-018-29516-1 PMID:30046160
- [7] M. J. Frisch, et al., Gaussian
16 Revision A.03, Gaussian Inc. Wallingford CT, 2013.
- [8] G. Landrum, et al.,
rdkit/rdkit: 2023_03_3 (Q1 2023) Release, Zenodo, 2023.
doi:10.5281/zenodo.8254217
- [9] T. Chen, C. Guestrin, XGBoost:
A Scalable Tree Boosting System, Proceedings of the 22nd ACM SIGKDD International
Conference on Knowledge Discovery and Data Mining, 2016, 785.
doi:10.1145/2939672.2939785
- [10] Y. Liu, K. W. Kong, D. T.
Wu, H. Y. Liu, H. B. Li, J. R. Zhang, et al., Food Chem., 374, 131635 (2022).
doi:10.1016/j.foodchem.2021.131635 PMID:34823934
- [11] A. Bellesia, E. Verzelloni,
D. Tagliazucchi, Int. J. Food Sci. Nutr., 66, 85 (2015). doi:10.3109/09637486.2014.953455
PMID:25519249
- [12] Y. Zhang, S. Cai, S. Ma, S.
Zhao, J. Yi, L. Zhou, Antioxidants, 11, 1873 (2022). doi:10.3390/antiox11101873
PMID:36290596