Abstract
While Sn-based perovskite solar cells have photoelectronic properties comparable to those
of lead halide perovskites, their low photoelectric conversion efficiency is a problem.
The main cause of this problem is the defect level caused by the presence of defects in
the crystal. In this study, we analyzed the defect structures in FASnI3 and
MASnI3 perovskites using first-principles calculations and focused on the
correlation between the photoelectric conversion efficiency and defect levels.
In both structures, the defect formation energy of VSn was low and tin tended
to be easily removed. In FASnI3, by changing the chemical potential to the
Sn-rich, I-poor condition, the defect levels that were easy to form in the Sn-poor, I-rich
condition became defect levels that were hard to form. It was also found that
MASnI3 has a wide range of thermodynamically stable regions
with no defect levels that are prone to form under any chemical potential condition.
Therefore, from the viewpoint of structural stability and structural defects, MA is
preferable to FA as the A-site cation of Sn-based perovskite.
1 Introduction
Perovskite solar cells, which use perovskite crystals in the light-absorbing layer, are
expected to become the next-generation solar cells because they have achieved high
photoelectric conversion efficiency comparable to that of silicon-based solar cells.
Currently, among perovskite solar cells, lead halide perovskite has achieved the highest
photoelectric conversion efficiency [1], but the
toxicity of lead has become a problem from the viewpoint of practical use, and lead-free
solar cells are required. Therefore, Sn-based perovskite solar cells are attracting
attention as a lead-free perovskite because of their high stability and resistance to heat
and light [2]. However, Sn-based perovskite solar
cells suffer from low photoelectric conversion efficiency. One of the main reasons for this
is "defects". The presence of defects in the crystal causes the appearance of deep levels in
the band gap (defect levels), which trap carriers and induce carrier recombination, leading
to low photoelectric conversion efficiency [3]. In
this study, we analyze the defect structures of FASnI3, which contains
formamidinium with high thermal stability at the A site, and MASnI3, which
contains methylammonium, the most widely studied, using first-principles calculations, and
discuss the influence of the A-site cations on the defect levels [4].
2 Methods
In FASnI3 and MASnI3, we focused on a series of point defects [5], namely atomic vacancies, self-interstitial atoms, and
anti-site defects, and considered the defect levels that are formed. First-principles
calculations were performed using the DFT calculation package VASP (Vienna ab
initio package) under the condition that the cutoff energy of the plane-wave
expansion is 520 eV. DFT calculations were performed using the PBE (Perdew-Burke-Ernzerhof)
exchange-correlation functional along with Grimme's D3 dispersion correction [6]. Defect structures for FASnI3 and
MASnI3 were created using 2×2×2 supercells containing 8 stoichiometric units.
The k-point sampling of the Brillouin zone was performed with a Γ-centered Monkhorst-Pack
scheme and determined to have a density of 2×2×2 grids for 2×2×2 supercells in the case of
defect structures and 2π×0.02 Å-1 for all other cases. The point-defect
calculations were conducted using Pydefect, a robust and open-source Python library [7].
3 Results and Discussion
We carried on structural optimization of FASnI3 and MASnI3 in
primitive cell. The space groups of FASnI3 and MASnI3 are
Amm2 and Pnma. The band gaps of FASnI3 and
MASnI3 are 0.60 eV and 0.70 eV, respectively, confirming a direct transition
band gap in both cases. The effective masses of holes and electrons were calculated to be
0.09 m0 and 0.214 m0 for
FASnI3 and 0.115 m0 and 0.101
m0 for MASnI3. These values are similar to the
results of a band gap of 0.65 eV with an effective mass of about 0.11
m0 and 0.28 m0 for
FASnI3 [8], and a band gap of about
0.4-0.8 eV with an effective mass of 0.101 m0 and 0.062
m0 for MASnI3 [9], respectively.
A chemical potential phase diagram is created by conditions under which FASnI3
and MASnI3 are thermodynamically stable. For example, in the case of drawing the
chemical potential phase diagram of FASnI3, we can calculate the formation energy
ΔHf of competing phases such as FA, Sn, I, FAI,
SnI2, and SnI4. Therefore, the condition under which
FASnI3 is thermodynamically stable [10]
is,
and
the conditions for not generating other competing phases such as FAI, SnI2 and
SnI4
are,
Similarly, for MASnI3, the formation energies ΔHf of
competing phases such as MA, Sn, I, MAI, SnI2, and SnI4 were
calculated. A phase diagram was prepared considering the conditions for thermodynamically
stable formation of MASnI3 and the conditions to avoid the formation of other
competing phases such as MAI, SnI2, and SnI4.
Figure 1 shows the chemical potential phase
diagram of (a) FASnI3 and (b) MASnI3. The horizontal axis is the
chemical potential of Sn, and the vertical axis is the chemical potential of I. The red area
indicates the stable region of FASnI3 and MASnI3. Compared to
MASnI3, the region of FASnI3 is significantly narrower. In other
words, FASnI3 is more dependent on the chemical potential of the constituent
elements than MASnI3. Therefore, it was found that the crystal growth conditions
must be carefully controlled when synthesizing FASnI3.

Figure 2 shows the defect formation energies of
FASnI3. Defect levels that easily form are those with a valence change at a
defect formation energy of 0 eV or less. In particular, since defect levels appearing in the
band gap affect the photoelectric conversion efficiency, we focused on the defect formation
energy of defect levels appearing in the band gap. Under the Sn-poor, I-rich condition
(Figure 2 (a)), ISn(3-/1-) defect
levels, which are tin-depleted and iodine-infused defects, tend to form. However, in the
Sn-rich, I-poor condition (Figure 2 (b)), the
defect formation energy of ISn(3-/1-) is above 0 eV, indicating that this defect
level is difficult to form. Therefore, in the Sn-rich, I-poor condition, there is not a
defect level that is easy to form. On the other hand, MASnI3 does not show any
defect levels that affect the photoelectric conversion efficiency, because no levels are
easily formed under any growth conditions, including the Sn-poor, I-rich condition (Figure 3 (a)), the Sn-rich, I-poor condition (Figure 3 (b)).

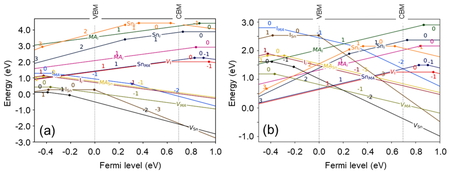
Figure 4 shows the defect level (thermodynamic
ionization levels) diagrams for each structure; for FASnI3, eight defect levels
appear in the band gap. However, except for ISn(3-/1-),the seven defect levels
are difficult to form under all growth conditions, and ISn(3-/1-) is also
difficult to form under Sn-rich and I-poor conditions. In MASnI3, six defect
levels were found in the band gap, all of which are difficult to form under any growth
conditions. In all structures, the VSn defect formation energy was found to be
low and tin tends to be easily released. This may be attributed to the antibonding nature of
the Sn-5s and I-5p orbitals at the upper end of the conduction band in each structure.
4 Conclusion
From the above results, we may conclude that the formation of defect levels can be
controlled in FASnI3 by setting Sn-rich, I-poor conditions, which is expected to
improve the photoelectric conversion efficiency. In MASnI3, it was found that
defect levels were less likely to form in the bandgap under any conditions, and that
thermodynamically stable range for equilibrium growth was a wider than that of
FASnI3. Therefore, in terms of structural stability and strong defect
resistance, MA is considered a more suitable A-site cation for tin perovskite solar cell
materials than FA.
Acknowledgement
This research is supported by NEDO project of Research and Development Program for
Promoting Innovative Clean Energy Technologies Through International Collaboration. We are
very grateful to Prof. Shuji Hayase, University of Electro-Communications, for his helpful
discussions. The DFT calculations were carried out at the Research Center for Computational
Science, Okazaki, Japan (Project: 22-IMS-C064) and the Center for Computational Materials
Science, Institute for Materials Research, Tohoku University for the use of MASAMUNE-IMR,
(Project No. 202303-SCKXX-0025).
References
- [1]Best Research-Cell
Efficiency Chart | Photovoltaic Research | NREL, (n.d.).
- [2] G. Nasti, A. Abate, Adv.
Energy Mater., 10, 1902467 (2020). doi:10.1002/aenm.201902467
- [3] D. Meggiolaro, D.
Ricciarelli, A. A. Alasmari, F. A. S. Alasmary, F. De Angelis, J. Phys. Chem. Lett., 11,
3546 (2020). doi:10.1021/acs.jpclett.0c00725 PMID:32298590
- [4] N. Liu, C. Yam, Phys. Chem.
Chem. Phys., 20, 6800 (2018). doi:10.1039/C8CP00280K PMID:29473061
- [5] I. Yonenaga, OYO BUTURI, 86,
1040 (2017).
- [6] S. Grimme, J. Antony, S.
Ehrlich, H. Krieg, J. Chem. Phys., 132, 154104 (2010). doi:10.1063/1.3382344
PMID:20423165
- [7] Y. Kumagai, N. Tsunoda, A.
Takahashi, F. Oba, Phys. Rev. Mater., 5, 123803 (2021).
doi:10.1103/PhysRevMaterials.5.123803
- [8] M. Roknuzzaman, J. A.
Alarco, H. Wang, A. Du, T. Tesfamichael, K. K. Ostrikov, Comput. Mater. Sci., 169, 109118
(2019). doi:10.1016/j.commatsci.2019.109118
- [9] M. S. Ozório, M. Srikanth,
R. Besse, J. L. F. Da Silva, Phys. Chem. Chem. Phys., 23, 2286 (2021).
doi:10.1039/D0CP06090A PMID:33443529
- [10] T. Shi, H. S. Zhang, W.
Meng, Q. Teng, M. Liu, X. Yang, et al., J. Mater. Chem. A Mater. Energy Sustain., 5, 15124
(2017). doi:10.1039/C7TA02662E