Abstract
Molecular dynamics (MD) simulations, accelerated by a universal neural network potential,
were employed to investigate the dynamic behaviors of ten inorganic anions
(Br-, Cl-, F-, OH-,
NO3-,
H2PO2-,
H2PO3-,
HPO42-, CO32- and
SO42-) intercalated into NiFe layered double hydroxide at varying
hydration levels. Our results show that the lattice parameters along the layered double
hydroxide (LDH) layers are minimally affected by the intercalated anions and water
content, while the lattice parameter perpendicular to the layers, i.e., the basal spacing,
is strongly influenced by the type of anion and hydration level. The basal spacing is
closely correlated with the ionic radius and charge of the anions, as well as the amount
of water uptake. Mean squared displacement (MSD) analysis reveals distinct behaviors of
the anions under different hydration conditions. While some anions, such as
NO3- and H2PO2-, exhibit
noticeable mobility, others remain largely immobilized, primarily due to strong
electrostatic interactions with the LDH layers and water molecules. Hydration weakens the
interaction between anions and the LDH but also restricts the mobility of anions due to
the formation of hydration shells. These findings provide insights into anion dynamics and
selectivity in NiFe LDH, which are critical for designing materials for anion removal
applications.
1 Introduction
Layered double hydroxides, with the formula
[M2+1-xM3+x(OH)2]x+(An-)x/n·yH2O,
consist of positively charged hydroxide layers where metal cations occupy octahedral sites,
while charge-balancing anions and water reside in the interlayer galleries (Fig. 1a). This structural flexibility allows LDHs to
host a wide range of anions, making them highly effective in applications such as
photocatalysis, drug delivery, and environmental remediation [1]. Among the various LDH compounds, NiFe-based LDHs stand out for their ease of
synthesis and tunable trivalent metal content or incorporation of other metals [2]. Importantly, NiFe LDHs exhibit excellent catalytic
activity, particularly in oxygen evolution reactions, and possess outstanding anion-exchange
capabilities [3, 4]. These features make them highly suitable for applications focused on the
selective removal of toxic anions from aqueous environments. However, the detailed
mechanisms governing anion interactions within their structure remain insufficiently
understood. Gaining deeper insight into these interactions is essential for optimizing
anion-exchange properties and enhancing their effectiveness in practical applications. For
this, we employ molecular dynamics simulations accelerated by machine learning interatomic
potentials to investigate how various anions interact when intercalated into the LDH. In
this work, 10 commonly studied monovalent and divalent inorganic anions are selected. They
are halides (Br-, Cl-, F-), hydroxide (OH-),
nitrate (NO3-), hypophosphite
(H2PO2-), phosphite
(H2PO3-), phosphate (HPO42-),
carbonate (CO32-) and sulfate (SO42-).

2 Methodology
We used a binary NiFe layered double hydroxide model with a Ni2+/Fe3+
ratio of 2:1 (x = 1/3). Total energy calculations were performed using a universal neural
network potential, namely the Preferred Potential (PFP) [5]. The choice of PFP in this study was motivated by its universal applicability
to diverse elements (supporting 96 naturally occurring elements from the periodic table) and
its ability to predict charge distributions, making it particularly well-suited for
simulating ionic systems such as the NiFe LDH investigated here. Furthermore, PFP has been
extensively tested in various applications, demonstrating its ability to achieve reasonable
accuracy across a wide range of material systems, including layered structures [6, 7],
multi-elemental nanoparticles and bulk structures [8,
9], gas molecule adsorption on nanoparticles [9, 10], and,
particularly, the dynamics of ionic systems [11]. In
fact, the suitability of PFP for modeling the NiFe LDH system was validated by comparing our
results, including basal spacing and X-ray Diffraction (XRD) patterns, with experimental
data. As shown in Table S1 and Fig. S1 in the Supplementary Information, good agreement was
found. In this work, PFP version 5.0.0 was used. PFP offers several calculation modes, which
support both molecular and crystal systems, with options to include or exclude Hubbard
corrections and dispersion corrections. For the case of NiFe, the mode for crystal systems
with Hubbard and dispersion corrections (CRYSTAL_PLUS_D3) was used to account for 3d
transition metals and layered structure. It is worth noting that in the calculations of
total energies using PFP with Hubbard correction, the dataset for training the potential was
prepared by applying the correction method by Dudarev et al. [12] with effective U values of 6.2 and 5.3 eV for Ni and
Fe, respectively. On the other hand, for the PFP mode including dispersion correction, the
DFT+D3 method by Grimme et al. [13,
14] was used for dealing with the dispersion
interaction. Initially, Ni and Fe were placed randomly on the cation sites of LDH layers.
The optimal atomic arrangement of Ni and Fe was then optimized using a simulated annealing
Monte Carlo method [6]. The results revealed a strong
preference for ordering, where Fe atoms tend to arrange in a
3a0×3a0
pattern (Fig. 1b) with
a0
is the lattice parameter of original LDH cell (space group
R3¯m)
along the
a0→
and
b0→
lattice vectors
(a0→ =b0→ =a0).
The arrangement of divalent (Ni2+) and trivalent (Fe3+) cations in the
LDH layers in this pattern is understandable because this kind of pattern minimizes the
electrostatic interaction within the layers. The ordering of Ni2+ and
Fe3+ cations is similar to the pattern proposed by experiments for the case of
MgAl LDH with Mg2+:Al3+ ratio of 2:1 [15]. With this stable arrangement, a
33×33×1
supercell consisting of 54 Ni atoms and 27 Fe atoms and a
43×33×1
supercell containing 72 Ni and 36 Fe atoms were constructed from the original LDH cell to
accommodate monovalent and divalent anions, respectively. The number of monovalent and
divalent anions intercalated into the LDH interlayers is 27 and 18, respectively, ensuring
the neutral charge state of the supercells. To investigate the effect of water on the
dynamics of anions and their interaction with the LDH, molecular dynamics simulations were
performed using PFP. Various hydration levels were examined with water-to-anion ratios of
0:1, 1:1, 2:1, and 3:1, corresponding to anhydrous state and hydrous states with number of
water molecules equal to, double, and triple the number of anions, respectively. Initially,
anions and water molecules were randomly distributed within the LDH interlayers. For the MD
simulations, the protocol began with a 0.1 ns NVE run, followed by a 0.4 ns
NVT ensemble run to equilibrate the system at 300 K. Subsequently, a 0.4
ns NPT run adjusted the system to 1 atm while maintaining 300 K. During the
NPT stage, density convergence was monitored, and lattice parameters were
estimated. Finally, a 0.2 ns production run in the NVT ensemble completed
the simulation process. A time step of 1 fs was used for all ensembles.
3 Results and Discussion
The calculated lattice parameters for the NiFe LDH cell along the
a→
and
b→
lattice vectors ranging from 3.129 to 3.162 Å, depending on the intercalated anions. These
values are close to those obtained from first-principles calculations (for the
CO32- anion) and experimental studies [16,17,18]. The lattice parameters of the LDH along these two lattice vectors
show minimal variation, regardless of the intercalated anions. However, as shown in Fig. 2, the lattice parameter along the
c→
lattice vector, and consequently the basal spacing, strongly depends on the type of
intercalated anions and the water content. A strong correlation is observed between the LDH
basal spacing and the ionic radii and charge states of the anions. Larger anion radii result
in increased basal spacing. Conversely, anions with higher formal charges lead to smaller
basal spacing due to stronger electrostatic interactions between the anions and the LDH
layers, pulling the layers closer together. Additionally, we can see that, when the
hydration level increases, the basal spacing also increases. To further understand the
interaction of anions with LDH and water, the Bader charge, an output of the PFP
calculations, was estimated for all investigated anions and is presented in Table 1.

Table 1. Average Bader charges on the components of anions at different water levels. X
denotes the element that differs from oxygen and hydrogen in the anions.
| Anion |
An-:H2O = 1:0 |
An-:H2O = 1:1 |
An-:H2O = 1:2 |
An-:H2O = 1:3 |
|
X |
O |
H |
X |
O |
H |
X |
O |
H |
X |
O |
H |
|
Br-
|
-0.718 |
|
|
-0.701 |
|
|
-0.704 |
|
|
-0.702 |
|
|
|
Cl-
|
-0.758 |
|
|
-0.730 |
|
|
-0.677 |
|
|
-0.698 |
|
|
|
F-
|
-0.942 |
|
|
-0.886 |
|
|
-0.866 |
|
|
-0.844 |
|
|
|
OH-
|
|
-1.323 |
0.626 |
|
-1.332 |
0.672 |
|
-1.315 |
0.677 |
|
-1.303 |
0.661 |
|
NO3-
|
0.831 |
-0.619 |
|
0.825 |
-0.601 |
|
0.827 |
-0.585 |
|
0.816 |
-0.577 |
|
|
H2PO2-
|
3.069 |
-1.625 |
-0.362 |
3.092 |
-1.617 |
-0.347 |
3.087 |
-1.620 |
-0.336 |
3.097 |
-1.620 |
-0.330 |
|
H2PO3-
|
3.458 |
-1.569 |
0.176 |
3.459 |
-1.556 |
0.177 |
3.458 |
-1.553 |
0.175 |
3.467 |
-1.554 |
0.171 |
|
CO32-
|
1.989 |
-1.211 |
|
1.969 |
-1.210 |
|
1.984 |
-1.206 |
|
1.995 |
-1.208 |
|
|
SO42-
|
3.757 |
-1.418 |
|
3.790 |
-1.407 |
|
3.819 |
-1.395 |
|
3.820 |
-1.387 |
|
|
HPO42-
|
3.712 |
-1.509 |
0.642 |
3.727 |
-1.504 |
0.678 |
3.730 |
-1.496 |
0.689 |
3.735 |
-1.489 |
0.666 |
Fig. 3a presents mean-squared displacement for
F- anions, with data shown for both anhydrous and hydrous states. We can see
that in the anhydrous state, the MSD remained flat throughout the simulation, indicating
that F− exhibited very limited mobility. This suggests that F- may be
tightly bound to the LDH layers due to strong electrostatic interactions. In contrast, in
the hydrous state, a gradual increase of MSD over time is observed. This implies that water
weakens the interaction between F- and the hydroxide layers, allowing more free
movement within the interlayer. Fig. 3b shows that
in the anhydrous state, F- anions bind to the hydroxide groups of the LDH through
hydrogen bonds (H-bonds) and are distributed evenly within the LDH interlayer. This
arrangement, similar to that of trivalent cations (Fe3+), minimizes repulsion
among the F- anions themselves. In the hydrous state, when water molecules are
intercalated into the interlayer, the oxygen atoms in the water form H-bonds with the LDH
hydroxide groups, weakening the interaction between the F- anions and the LDH.
With a small amount of intercalated water, there is enough space between F-
anions. Therefore, the basal spacing slightly expands. However, as more water is introduced,
additional H-bonds form between water molecules and the LDH hydroxide groups, expanding the
basal spacing quickly. Simultaneously, F- anions and water molecules form large
hydration shells, further weakening the direct bonding between the F- anions and
the LDH. For other halide anions (Br-, Cl-),a similar distribution and
change in interlayer spacing were observed as in the case of F-. However, in the
anhydrous state, Br- and Cl- can more easily jump between sites due to
their weaker interaction with the LDH (see Fig. S2). When water is intercalated, the
mobility of Br- and Cl- decreases because the formation of hydration
shells creates a steric hindrance effect that restricts the movement of these anions.
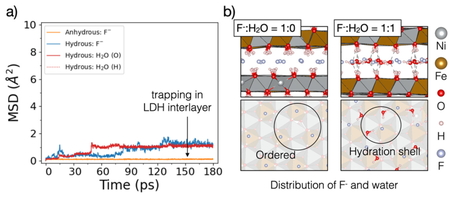
Fig. 4a shows that NO3-
exhibits significantly higher mobility compared to halides. This suggests weaker interaction
between NO3- and the LDH layers, allowing for more rapid diffusion. In
the anhydrous state, NO3- exhibits linear increases in MSD, indicating
diffusive behavior rather than jumping. In the hydrous state, the anion continued to show
linear increases in MSD, but at a slower rate compared to the dehydrated state. The mobility
of NO3- is reduced, likely due to the blocking of water that impedes
diffusion. The diffusion coefficient dropped from 3.396×10−6 cm2/s to
1.261×10−6 cm2/s. Fig. 4b
shows that unlike halides, the distribution of NO3- anions is not
strictly even within the LDH interlayer. This might be due to the delocalized negative
charge across the anion, as compared to F- or Cl-. In both anhydrous
and hydrous states, NO3- anion prefers a perpendicular or slightly
tilted orientation with the LDH layers. This orientation is similar to that observed from
experiment for the case of CaAl LDH [23]. By
comparing the interaction of NO3- and CO32-
(which share similar atomic conformation) with LDH, we found that NO3-
prefers a perpendicular or tilted orientation, while CO32- adopts a
planar orientation within the LDH interlayer. Notably, this planar orientation of
CO32- persists even as more water is introduced into the interlayer
(see Fig. S3). The difference arises from their geometry and charge:
NO3-, with its delocalized single negative charge (approximated from
ionic radius and Bader charge in Fig. 2 and Table 1), exhibits flexibility, favoring
perpendicular or tilted orientations due to weaker electrostatic interactions and versatile
hydrogen bonding. In contrast, CO32-,with its higher charge density,
strongly prefers an in-plane orientation, maximizing electrostatic attractions and hydrogen
bonding with LDH layers.

For the remaining anions, we found that H2PO2- behaves
similarly to NO3-, as it can diffuse within the LDH interlayer in the
anhydrous state. However, when water is present, the anion is heavily blocked by water and
becomes trapped in the LDH interlayer. From the estimation of the mean squared displacement
of all anions, we found that, in the hydrous state with a hydration level of
An−:H2O = 1:1, NiFe LDH has a high likelihood of selecting
OH-, SO42-, HPO42-,
CO32-, F-, H2PO3- from
water, while the selection of NO3- and
H2PO2- is less probable. This is qualitatively consistent
with experimental observations [24]. In general, the
observed differences in mobility between intercalated anions can be attributed to their
hydration shell characteristics, steric hindrance (due to the presence of water), and
competitive adsorption between the anions and water molecules. For instance, F-
forms a strong hydration shell, which moves along with the water molecules during diffusion,
allowing F- to exhibit increased mobility when water is introduced. In contrast,
NO3- has a larger ionic radius and experiences steric hindrance in
the presence of water, leading to reduced mobility. Additionally, for anions such as
Br- and Cl-, which also show changes in mobility upon hydration,
this can be explained by the competitive adsorption between the anions and water molecules,
as well as their weaker hydration shells compared to F-. Experimentally, the
hydration energies of Br-, Cl-, and F- are -104.0, -131.2,
and -278.8 kJ/mol, respectively [25].
To further understand the interactions among anions, water, and LDH, dispersion interaction
and short-range Coulomb interaction (modeled by the Yukawa potential) were analyzed. For the
dispersion interaction, additional calculations were performed using the CRYSTAL mode of the
potential and compared with data from the CRYSTAL_PLUS_D3 mode. We estimated that dispersion
interactions contribute about 2.2% to the total energy, while Coulomb interactions between
anions, water, and LDH (within a cutoff radius of 8 Å) account for approximately 7.0%. The
primary contribution likely comes from mixed ionic-covalent interactions within the LDH
layers and the hydrogen bonding network. Upon hydration, the dispersion energy slightly
increases, while the Coulomb interaction energy rises more significantly. Further
decomposition reveals that most of the Coulomb interaction comes from the interaction
between anions and LDH hydroxide groups, as well as between anions and water. As the
hydration level increases, the anion-LDH interaction weakens due to the formation of a
hydration shell around the anions, which blocks direct interaction with the LDH hydroxide
groups. Meanwhile, the interaction between anions and water increases (Fig. 5). Lastly, the hydrogen bonding network, particularly involving
water molecules, may play a significant role in the mobility and interaction of intercalated
anions with the LDH layers. Anions like F-, with a strong hydration shell, form
robust hydrogen bonds with water molecules, facilitating their movement during diffusion.
Similarly, Br- and Cl-, despite having weaker hydration shells
compared to F-, also form hydrogen bonds with water, allowing for increased
mobility upon hydration, albeit to a lesser extent. Although not discussed in detail here,
this aspect warrants further investigation in future studies.

4 Conclusion
We performed molecular dynamics simulations using a universal neural network potential to
study the dynamic behaviors of ten inorganic anions (Br-, Cl-,
F-, OH-, NO3-,
H2PO2-,
H2PO3-,
HPO42-, CO32- and
SO42-) intercalated into NiFe layered double hydroxide
([Ni2/3Fe1/3(OH)2]1/3+(An-)1/3n)
at different hydration levels. First, we found that while the lattice parameters along the
LDH layers are less sensitive to the intercalation of different anions and water levels, the
lattice parameter perpendicular to the LDH layers is strongly influenced by the type of
intercalated anions and the amount of water. The basal spacing of the LDH is closely
correlated with the anion's ionic radius and charge state. Second, the mean squared
displacement analysis reveals distinct behaviors for selected anions under varying hydration
conditions, showing differences in mobility and interactions within the NiFe LDH structure.
In the short-time MD simulations, the diffusive behavior of all anions, except
NO3- and H2PO2-, was barely
observed. These anions were mostly trapped in the interlayer due to their strong
electrostatic interactions with the LDH hydroxide groups or surrounding water. Specifically,
the presence of water significantly hindered the movement of the anions. Further analysis
indicates that the majority of the Coulomb interactions stem from interactions between
anions and the LDH hydroxide groups, as well as between anions and water. For future work,
longer simulation times could be employed to better capture the anion selectivity and
provide deeper insights into the stability and dynamics of intercalated anions over extended
periods. Additionally, exploring the effects of varying ionic strengths and mixed-ion
intercalation may further enhance the understanding of ion dynamics in LDH materials.
Acknowledgement
This work was supported by Council for Science, Technology and Innovation (CSTI),
Cross-ministerial Strategic Innovation Promotion Program (SIP), the 3rd period of
SIP "Creating a materials innovation ecosystem for industrialization".
References
- [1] L. Dang, H. Liang, J. Zhuo,
B. K. Lamb, H. Sheng, Y. Yang, et al., Chem. Mater., 30, 4321 (2018). and references
thereinhttps://doi.org/10.1021/acs.chemmater.8b01334
- [2] M. Tipplook, T. Sudare, H.
Shiiba, A. Seki, K. Teshima, ACS Appl. Mater. Interfaces, 13, 51186 (2021).
https://doi.org/10.1021/acsami.1c13706 PMID:34672191
- [3] F. Liao, X. Zhao, G. Yang,
Q. Cheng, L. Mao, L. Chen, J. Alloys Compd., 872, 159649 (2021).
https://doi.org/10.1016/j.jallcom.2021.159649
- [4] Q. Wang, D. O’Hare, Chem.
Rev., 112, 4124 (2012). https://doi.org/10.1021/cr200434v PMID:22452296
- [5] S. Takamoto, C. Shinagawa,
D. Motoki, K. Nakago, W. Li, I. Kurata, et al., Nat. Commun., 13, 2991 (2022).
https://doi.org/10.1038/s41467-022-30687-9 PMID:35637178
- [6] T. Q. Nguyen, M. Koyama, (2023,
December 11-16). Global Optimization of Atomic Arrangement in Multicomponent Alloys
Accelerated by Universal Neural Network Potential [Poster session]. MRM2023/IUMRS-ICA2023
Grand Meeting, Kyoto, Japan.
- [7] T. Chen, E. H. Otal, T. Q.
Nguyen, S. Narumi, M. Koyama, N. Zettsu, Chem. Mater., 37, 709 (2025).
https://doi.org/10.1021/acs.chemmater.4c02852
- [8] T. Q. Nguyen, Y. Nanba, M.
Koyama, J. Comput. Chem. Jpn., 21, 111 (2022).
https://doi.org/10.2477/jccj.2023-0015
- [9] S. M. Aspera, G. V. Huerta,
Y. Nanba, K. Hisama, M. Koyama, J. Comput. Chem. Jpn., 23, 19 (2024).
https://doi.org/10.2477/jccj.2024-0006
- [10] A. Tamura, G. Valadez
Huerta, Y. Nanba, K. Hisama, M. Koyama, J. Comput. Chem. Jpn., 21, 129 (2022).
https://doi.org/10.2477/jccj.2023-0016
- [11] K. Hisama, G. Valadez
Huerta, M. Koyama, Comput. Mater. Sci., 218, 111955 (2023).
https://doi.org/10.1016/j.commatsci.2022.111955
- [12] S. L. Dudarev, G. A. Botton,
S. Y. Savrasov, C. J. Humphreys, A. P. Sutton, Phys. Rev. B Condens. Matter, 57, 1505
(1998). https://doi.org/10.1103/PhysRevB.57.1505
- [13] S. Grimme, J. Antony, S.
Ehrlich, H. Krieg, J. Chem. Phys., 132, 154104 (2010). https://doi.org/10.1063/1.3382344
PMID:20423165
- [14] S. Grimme, S. Ehrlich, L.
Goerigk, J. Comput. Chem., 32, 1456 (2011).
https://doi.org/10.1002/jcc.21759
- [15] S. V. Krivovichev, V. N.
Yakovenchuk, A. A. Zolotarev, Jr., G. N. Ivanyuk, Y. A. Pakhomovsky, Chimia (Aarau), 64,
730 (2010). https://doi.org/10.2533/chimia.2010.730 PMID:21138162
- [16] Y. Dong, P. Zhang, Y. Kou,
Z. Yang, Y. Li, X. Sun, Catal. Lett., 145, 1541 (2015).
https://doi.org/10.1007/s10562-015-1561-0
- [17] M. del Arco, P. Malet, R.
Trujillano, V. Rives, Chem. Mater., 11, 624 (1999).
https://doi.org/10.1021/cm9804923
- [18] S. Abelló, E. Bolshak, D.
Montané, Appl. Catal. A Gen., 450, 261 (2013).
https://doi.org/10.1016/j.apcata.2012.10.035
- [19] R. D. Shannon, Acta
Crystallogr. A, 32, 751 (1976). https://doi.org/10.1107/S0567739476001551
- [20] Y. Marcus, J. Chem. Phys.,
137, 154501 (2012). https://doi.org/10.1063/1.4758071 PMID:23083175
- [21] I. Louati, F. Guesmi, A.
Chaabouni, C. Hannachi, B. Hamrouni, Water, Qual. Res. J., 51, 60 (2016).
- [22] H. D. B. Jenkins, K. P.
Thakur, J. Chem. Educ., 56, 576 (1979). https://doi.org/10.1021/ed056p576
- [23] A. N. Narayanappa, S.
Nagendran, P. V. Kamath, New J. Chem., 45, 5837 (2021).
https://doi.org/10.1039/D1NJ00148E
- [24] We compared our findings with
those of F. Hayashi of Shinshu University (personal communication, 2024), who reported
similar results in their ongoing research on NiFe LDH.
- [25] W. M. Haynes, ed., CRC Handbook
of Chemistry and Physics, 97th ed., (Boca Raton: CRC Press, 2016), 5-65.