Review
A New Synthetic Tool: The Pseudo-Intramolecular Process
2018 Volume 67 Issue 1 Pages 11-19

Details
2018 Volume 67 Issue 1 Pages 11-19
An efficient synthetic protocol based on a new concept named “the pseudo-intramolecular process” is developed. Substrates are brought closed to each other by forming a salt, and this spatial proximity facilitates an efficient reaction like an intramolecular process, despite actually proceeding via an intermolecular pathway. This concept can be widely applied in organic synthesis. For instance, α-aryl-β-keto esters undergo a transacylation with amines accompanied by high efficiency, regioselectivity, and chemoselectivity. On the other hand, α-nitro-β-keto nitriles react via a tandem cyclization to afford polyfunctionalized azaheterocyclic compounds, which cannot be easily prepared by alternative methods. These synthetic protocols are practically useful because each reaction can be conducted without using any special reagent under mild reaction conditions through only simple experimental manipulations.
Organic reactions usually proceed via an intermolecular process, in which two reactants should encounter each other within a reaction mixture and collide at a suitable angle and with sufficient energy (Scheme 1-a). In contrast, intramolecular reactions occur faster than intermolecular reactions because of the high collision frequency of the reaction sites, which can be attributed to their spatial proximity (Scheme 1-b). In this case, even a less nucleophilic site such as a neutral atom can react efficiently. In other words, the efficiency of a reaction can be improved even in the case of an intermolecular process when the collision frequency of the reactants is increased. Indeed, the employment of reaction fields, such as capsules, cages, bowls, and micelles has been recognized as a useful method in organic synthesis to bring the reactants close to one another, so that the reaction proceeds rapidly to afford the product1),2). However, it is still difficult to achieve a high reaction efficiency under mild conditions without using any additional contrivance, such as a reaction field, catalyst, or activating agent. In this context, the development of an efficient protocol remains one of the greatest challenges.
We demonstrated a conceptually novel reaction that involves a pseudo-intramolecular process, which proceeds like an intramolecular process although it is actually an intermolecular process. The general model of this pseudo-intramolecular process is illustrated in Scheme 1-c3). First, an acidic hydrogen attracts an amine by forming an ammonium salt. When the nucleophilic amine is liberated under equilibrium, it comes in close proximity to the electrophilic substrate, forming an intimate pair. This spatial proximity enables an efficient reaction to take place. This new concept facilitates the synthesis of polyfunctionalized compounds, which is described in this review.

Concepts of the pseudo-intramolecular process.
2.1 Acyl group activation by α-arylation of β-keto esters
While α-arylated β-diketones are commonly used as bidentate ligands and a synthetic reagents4),5)6), α-arylated β-keto esters are less popular compounds because the C-C bond can be easily cleaved during the copper catalyzed coupling reaction of ethyl acetoacetate with aryl iodides (Scheme 2-a)7). In the course of our study, we also found that deacylation easily occurs upon treatment of α-quinolyl-β-keto esters with hydrazine or silica gel (Scheme 2-b)8). These experimental results clearly reveal that the acyl group of a β-keto ester can be activated by the introduction of an α-aryl group. This observation prompted us to activate the acyl group of a β-keto ester with an α-DNP (2,4-dinitrophenyl) group, which can be easily introduced by nucleophilic substitution (Scheme 2-c).

Deacylation of α-arylated β-keto esters.
The treatment of ethyl acetoacetate with propylamine 2a did not lead to the formation of any product. On the contrary, α-arylated keto ester 1 underwent transacylation at room temperature to afford N-propylacetamide 3a and ethyl phenylacetate 4, quantitatively (Table 1, run 1) 9),10). Although the transacylation was also applicable to isopropylamine 2b (run 2), the reaction was found to be considerably affected by the amine steric bulk. While 90% of 1 was consumed at room temperature within 4 h when reacted with propylamine 2a, a reaction time of 15 h r was needed to obtain a 90% conversion in the case of 2b. In the presence of tert-butylamine 2c, the transacylation did not occur even at 140°C in a sealed tube (run 3). Aniline 2d also did not furnish any transacylated products 3d and 4 (run 4). Although 2c and 2d did not undergo transacylation, a different reactivity was observed in these cases. When aliphatic amine 2c was added to a solution of 1, ammonium enolate 5c was immediately formed; however, no formation of anilinium enolate 5d was observed in the case of the less basic compound 2d. In this case, the addition of triethylamine to convert 1 to the corresponding ammonium enolate was effective to produce the transacylation. However, extended reaction times and a higher reaction temperature were required (run 5).

Transacylation of keto ester 1 with amines 2.
The transacylation is assumed to proceed as shown in Scheme 3. Owing to steric repulsion, the DNP and keto ester moieties are twisted with respect to each other at an angle of ca. 60°11). When a sterically hindered substituent is introduced at the α-position to the carbonyl group, the keto form using an sp3 orbital is destabilized giving a stable enol using an sp2 orbital12). Indeed, only the signals corresponding to the enol form were observed in the 1H NMR spectrum of 1. The acidity of ethyl acetoacetate increases by approximately 7 orders of magnitude by introducing a benzene ring because the O-H proton is more acidic than the C-H13). As a result, keto ester 1 becomes a stronger acid than acetic acid. Hence, upon addition of an amine, the corresponding ammonium enolate 5 is immediately formed, accompanied by the fission of an intramolecular hydrogen bond. The formation of the keto form under equilibrium conditions brings the nucleophile and electrophile in close proximity giving rise to an intimate-pair 6; this results in a quantitative transacylation under mild conditions, which is reminiscent of an intramolecular process even though the reaction actually follows an intermolecular pathway. Thus, it is a pseudo-intramolecular process. The less basic aniline 2d is not attracted to keto ester 1, which was not able to undergo the pseudo-intramolecular process. Although the acyl transfer from 1 to 2d occurs upon addition of triethylamine, the reaction rate is quite slow since it is an intermolecular process.

A plausible mechanism for the transacylation.
NMR studies helped to confirm that this transacylation reaction proceeds via a pseudo-intramolecular process14). Solvation is one of the crucial factors affecting the reactivity of an intermolecular process, because the collision frequency is diminished when two substrates are solvated15). As a result, the rate of an intermolecular process changes depending on the polarity of the reaction medium. On the other hand, for an intramolecular process the reaction sites are close to each other, thus they should be less influenced by the polarity of the solvent16). The transacylation was monitored by 1H NMR spectroscopy using three deuterated solvents (acetonitrile-d3, THF-d8, and benzene-d6) having different dielectric constants (εr) and dipole moments (μ)17). It was found that the transacylation proceeded with the same reaction rates despite the extremely different polarities of these solvents (Fig. 1). This result strongly indicates that the transacylation reaction was not affected by the polarity of the solvent. Furthermore, the reaction rate did not change significantly even when the reaction mixture was highly diluted, which is typical of an intramolecular process14). On the basis of the solvation and concentration results obtained by NMR studies, the transacylation was confirmed to proceed via a pseudo-intramolecular process.

Time/conversion curve for 5a in different solvents and rate constants k and relative rate constants krel with the solvent parameters.
2.2 Synthesis of unsymmetrical malonates9)
In order to demonstrate one of the potential synthetic applications of the transacylation reaction, the synthesis of unsymmetrical malonic acid derivatives was investigated using arylated acetonedicarboxylate 7 instead of acetoacetate 1. Dicarboxylic acids and their derivatives are important synthetic intermediates for the preparation of various types of polyfunctionalized systems. Among these, unsymmetrical acid derivatives are valuable tools for elaborate molecular design. While succinic (or glutaric) acid amide esters are easily prepared by aminolysis of the corresponding acid anhydrides, the synthesis of unsymmetrical malonates18),19) cannot be performed in a similar way since malonic anhydride is not a commonly used reagent. Hence, they are generally prepared by either aminolysis of diethyl malonate or chemical conversion of malonic acid. These methods require a selective chemical transformation of one of the two equivalent carbonyl groups, which sometimes is accompanied by low selectivity and may involve troublesome manipulations to achieve separation of the products.
Upon treatment with propylamine 2a, keto ester 7 underwent transacylation to quantitatively afford malonic acid amide ester 8a (Table 2, run 1). As observed for acetoacetate 1, the transacylation of 7 was sensitive to the steric bulk of the amines used (runs 1–3). During the reaction of 7 with several anilines, a significant substituent effect was observed; while unreacted p-nitroaniline 2e was recovered, even when the mixture was heated in the presence of triethylamine, the more basic p-methoxyaniline 2f effectively reacted at room temperature in the absence of triethylamine to afford amide ester 8f (runs 4–7). These experimental results indicate that the initial formation of the ammonium enolate 5 is crucial for an efficient transacylation to occur.

Synthesis of unsymmetrical malonic acid derivatives 8.
2.3 Regio- and chemoselective acylations9)
The transacylation is sensitive to the steric bulk of amine 2, which was estimated by a competition reaction. This reaction precisely recognized the presence of a methyl group at the α-position of the amino group (Table 3, run 1) ; the different location of a methyl group either at the α- or β-positions was also effectively distinguished (run 2). The presence of a methyl group at the β-position was also recognized, although the selectivity was lowered (run 3). The steric congestion of this intimate pair was responsible for the sensitivity of the reaction to the bulkiness of the nucleophile, thus enabling the regioselective acylation of 1,2-diaminopropane 2g (Table 4). In this reaction, only the terminal amino group was acylated without any modification of the internal amino group, which cannot be achieved by the acylation of 2g with commercially available acyl chloride. Furthermore, it was not necessary to use a base in order to trap the generated acid. This transacylation was also able to recognize the number of alkyl groups on the amino group; only the primary amino group of 2h was acylated without any reaction occurring at the secondary amino group (Scheme 4).

Estimation of sensitivity for steric bulk of amines 2 by competitive reaction.

Regioselective acylation of 1,2-diamine 2g.
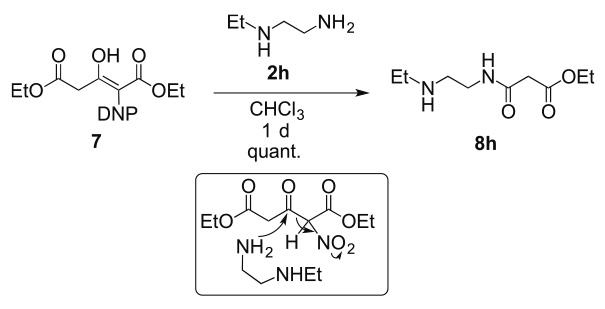
Recognition between primary and secondary amines 2h.
When ethanol 9i was used as nucleophile, the transacylation proceeded extremely slowly even in the presence of triethylamine under heating conditions due to the intermolecular process. On the other hand, when 2-(tert-butylamino) ethanol 9j was allowed to react with compound 1, an O-acylation proceeded to afford 10j without formation of any detectable N-acylated product 3j. For this reaction, the protection of the amino group was not necessary. It is noteworthy that the reaction rate of 9j was considerably higher than that of ethanol 9i in the presence of triethylamine (Table 5). In the case of 9j, the formation of ammonium enolate 5j is believed to facilitate the approach of the hydroxy group to the keto ester moiety, while, at the same time, the sterically hindered tert-butyl group prevents the reaction of the amino group with the keto ester to form the intimate pair 6j (Scheme 5). Hence, it can be concluded that this reaction proceeded via a pseudo-intramolecular process.

O-Acylation of amino alcohol 9j via the pseudo-intramolecular process.

A plausible mechanism for the O-acylation of 9j.
2.4 Transacylation using α-nitro-β-keto esters20)
As mentioned above, compounds possessing an acidic hydrogen and a functional group can serve as substrates in the pseudo-intramolecular process. A number of nitro compounds satisfy these criteria; for example, Ballini et al. observed an unusually high reactivity of α-nitroketones that efficiently underwent the deacylation21), which is considered to proceed via a pseudo-intramolecular pathway.
Nitration of diethyl acetonedicarboxylate was readily accomplished by Laikhter’s method22)using two-phase reaction conditions (dichloromethane/sulfuric acid) to afford α-nitrated keto ester 11. When a mixture of 11 and propylamine 2a in dichloromethane is stirred at room temperature for 1 day, the transacylation quantitatively proceeded to give amide ester 8a along with ethyl nitroacetate (Table 6, run 1). Since 11 exhibited a higher reactivity compared to α-arylated keto ester 1, bulkier amines such as 2b and 2c were also suit reaction partners affording the corresponding malonates 8b and 8c, respectively (runs 2 and 3). Since a strongly electron-withdrawing nitro group activates the adjacent carbonyl group more than a DNP group, alcohols 9 could also be employed as nucleophiles. When a solution of 11 in methanol 9k was stirred at room temperature, the unsymmetrical diester 12k was obtained in high yield (run 4). The amount of 9k could be decreased to 5 molar equivalents by conducting the reaction in a chloroform solution under reflux conditions (run 5). In addition, sterically hindered isopropyl alcohol 9l and tert-butyl alcohol 9m (in the presence of potassium tert-butoxide) could also be employed, yielding diesters 12l and 12m in moderate yields, respectively; however, these reactions proceeded slowly due to the intermolecular process (runs 6 and 7). Indeed, when the reactions between 11 and propylamine 2a /methanol 9k conducted at room temperature were monitored by 1H NMR, different behaviors were observed. In the case of 2a, the immediate formation of ammonium nitronate was observed, and the transacylation proceeded to completion within 3 h. The transacylationof 11 with methanol 9k also occurred, however, a reaction time for 30 h was required to achieve the consumption of 11. In summary, these results indicate that the former reaction occurred via a pseudo-intramolecular process through the formation of ammonium nitronate, while the latter reaction proceeded only slowly owing to the sluggishness of the intermolecular process, in addition to the low nucleophilicity of methanol 9k.

Transacylation of α-nitro-β-keto ester 11.
To confirm the accelerating effect of the intimate pair formation, we monitored the reaction of 2-(dimethylamino) ethanol 9n (124 mM) and ethanol 9o (124 mM) with 11 by 1H NMR spectroscopy, using chloroform-d as solvent at 24°C (Table 7). Both nucleophiles 9n and 9o underwent O-acylation with 11 to give the corresponding diesters 12n and 12o, respectively. In the case of aminoethanol 9n, the respective salt 13n immediately formed after the addition. A significant difference in the reactivity was observed between alcohols 9n and 9o. Although the reaction of 9n with 11 afforded 13n in 90% yield after 2 h, compound 13o was obtained in only 6% yield after the same reaction period (runs 1 and 3). The reactivity of ethanol 9o did not increase when triethylamine was added to the reaction mixture, supporting the assumption that the reaction of 9n proceeds via a pseudo-intramolecular process, in which an intimate pair is formed upon deprotonation of 11 by the dimethylamino group (Scheme 6). Further experiments under diluted conditions allowed us to determine whether the reaction proceeded via a pseudo-intramolecular or an intermolecular process. When the concentration of both 11 and 9o (62 mM) was decreased to half of its original value, the yield of 9o after 6 h substantially decreased from 17 to 3%, suggesting that the reaction proceeds via an intermolecular mechanism (runs 4 and 5). On the other hand, aminoalcohol 9n (62 mM) was readily converted to 12n (90% after 2 h) despite being diluted, supporting the pseudo-intramolecular process (run 2).

Comparison between the reactivity of aminoethanol 9l and ethanol 9m with 11.

A plausible mechanism for the O-acylation of aminoethanol 9l.
3.1 Synthesis of 1,4-dihydropyridines23),24)
In the pseudo-intramolecular process, the substrate requires an acidic hydrogen to recruit an amine as well as a second functional group. Since many compounds exist that satisfy these criteria, it is anticipated that the pseudo-intramolecular process can be applied to general organic synthesis. In the course of our study on the chemical conversions of the pyridinium salt of nitroisoxazolone 1425),26), α-nitro-δ-keto nitrile1527)was found to satisfy the two criteria required for a compound to act as substrate in the pseudo-intramolecular reaction (Scheme 7).

Synthesis of α-nitro-β-keto nitrile 15.
Thus, ammonium salt 16a was quantitatively formed upon treatment of keto nitrile 15 with propylamine 2a. When a solution of 16a in acetonitrile was heated to reflux, the vicinally functionalized 1,4-dihydropyridine 17a was isolated in 71% yield (Table 8, run 1). An intimate pair was also formed by liberating the amine under equilibrium conditions upon heating ammonium salt 16a. The amine attacked the proximate cyano group, which then reacted with the acyl group to form a six-membered ring. Subsequent dehydration and proton transfer led to the formation of dihydropyridine 17a (Scheme 8). This reaction was not applicable to amines 2b–d possessing a bulky group because of the congestion around the reaction sites of the intimate pair. (runs 2–4). Insertion of a methylene group, which acts as a spacer between the bulky group and the amino functional group, facilitated the cyclization to afford dihydropyridines 17p–t (runs 5–9). When keto nitrile 18, which does not have two methyl groups at the β-position, was allowed to react with benzylamine 2s under somewhat severe conditions, air oxidation occurs to afford aromatized pyridine 19 in 63% yield (Scheme 9)28).

Synthesis of difunctionalized 1,4-dihydropyridines 17.

A plausible mechanism for the formation of dihydropyridines 17.

Synthesis of difunstionalized pyridine 19.
3.2 Synthesis of diazabicylic compounds23),29)
α-Nitro-δ-keto nitrile 15 exhibits multiple functionalities. When a dinucleophilic diamine 20 is employed instead of monoamine 2, both nucleophilic and electrophilic sites remain within a spatial proximity, even after the formation of ammonium salt 21. These sites may readily react to afford a different type of framework. As illustrated in Scheme 10 for 1,2-diaminoethane 20a, the reaction is initiated by a deprotonation at the α--carbon of δ-keto nitrile 15 to afford ammonium salt 21a, which brings the two reactants, 15 and 20a in proximity, giving rise to a pseudo-intramolecular imination that yields zwitterionic imine 22a30). When imine 22a is heated, a small quantity of amine 23a is formed under the equilibrium conditions, and the nucleophilic and electrophilic character of the amino and cyano group, respectively, are reversed. As a result, two rings are simultaneously constructed by the nucleophilic attack of the amino group on the imino carbon, followed by the subsequent attack of the imino nitrogen on the cyano group, yielding bicyclic product 24a in 76% yield (Table 9, run 1). In order to confirm that this reaction proceeds via a pseudo-intramolecular process, δ-keto nitrile 25, which does not carry a nitro group, was subjected to the reaction with diamine 23a under the same reaction conditions; however, only unreacted 25 was recovered (Scheme 11). Owing to the presence of three carbons between the two functional groups (cyano and keto groups) in 25, the nitro group cannot activate the carbonyl group by an electron-withdrawing inductive effect. Therefore, the main role of the nitro group is to make the α-hydrogen more acidic so that it can interact with diamine 23a to form ammonium salt 21a, which is an essential step to trigger the pseudo-intramolecular process, as shown in Scheme 10.

A plausible mechanism for the formation of diazabicyclic compound 23.

Synthesis of diazabicyclic compounds 24 and 26.

Reaction of keto nitrile 25 with diamine 20a.
The formation of 26 from 18 via bicyclization indicates that the presence of two methyl groups at the β-position of the δ-keto nitrile is not required for this reaction (run 2). When 1,2-diaminopropane 20b was used, an equimolar mixture of regioisomers 24b and 24b’ was formed, indicating that the initial imination was unaffected by the presence of a methyl group on the ethylene chain (run 3). The reactivity of diamine 20c, which bears an N-ethyl group, was similar to that of unsubstituted diamine 30a, and a bicyclic product 24c was obtained, leading us to conclude that the N-ethyl group did not interfere with the reaction (run 4). This is noteworthy because it suggests that, in the present reaction, the primary amino group is preferred over the secondary, even though they display similar basicity. Furthermore, it was possible to construct relatively large condensed rings by employing diamines 20d and 20e. Therefore, in the presence of 1,3-diaminopropane 20d, the bicyclization proceeded to afford diazabicyclo [4.4.0] decane 24d in 85% yield (run 5). The use of 1,4-diaminobutane 20e resulted in the formation of a seven-membered ring (run 6).
α-Nitro-δ-keto ester 27 also satisfies the two criteria required to act as a substrate in the pseudo-intramolecular process; this compound has both an acidic hydrogen and functional groups. Thus, keto ester 27 served as substrate in this process to afford two isomeric products 28 and 29 that were isolated in 74% and 10% yields, respectively, as a result of the reaction with 1,2-diaminoethane 20a (Scheme 12)31).

Synthesis of two isomeric α-nitro bicyclic lactones 28 and 29.
A conceptually novel reaction involving a pseudo-intramolecular process was developed and applied to the synthesis of polyfunctionalized heterocyclic compounds. Substrates that satisfied the two criteria, i.e., the presence of an acidic hydrogen and additional functional group (s), could undergo this process efficiently. These requirements do not pose strict limitations to the substrate’s molecular design and synthesis.
When a suitable substrate was allowed to react with an amine, an intimate pair formed via an intermediate ammonium salt, owing to the proximity between the nucleophilic amine and the electrophilic substrate. This spatial proximity enables an efficient reaction to proceed similarly to an intramolecular process, even though it actually is an intramolecular. Indeed, the reaction efficiently occurs under mild conditions without formation of considerable amounts of by-products. Furthermore, the steric congestion of the intimate pair makes the reaction sensitive to the bulkiness of the nucleophile, which allows the reactions to be conducted in a regio- and chemoselective manner.
The present method can be performed using simple manipulations, under air, without being affected by moisture, and affords multiply functionalized compounds that can be further converted into many new compounds. Hence, this pseudo-intramolecular reaction is expected to emerge as a powerful tool in the synthesis of polyfunctionalized compounds that cannot be prepared by alternative methods.
ACKNOWLEDGMENT
I am grateful for collaboration with Prof. Masahiro Ariga and Prof. Yasuo Tohda (Osaka Kyoiku Univ.), Prof. Yoshito Tobe and Prof. Shinobu Itoh (Osaka Univ.), Prof. Kazuhiko Saigo, Prof. Kazuya Kobiro, Dr. Shotaro Hirao and Dr. Haruyasu Asahara (Kochi Univ. Tech.). I also acknowledge my students for their sincere works for the chemistry.