The Inter-College of Physical Chemistry 2019
Controlled Self-Assembly of Amphiphilic Random Copolymers into Folded Micelles and Nanostructure Materials
2020 Volume 69 Issue 6 Pages 529-538

Details
2020 Volume 69 Issue 6 Pages 529-538
In this review, we report controlled self-assembly systems of amphiphilic random copolymers in aqueous or organic media and the solid state to produce folded micelles, related nanoaggregates, vesicles, and microphase separation materials. The key features of random copolymer self-assemblies are 1) self-folding of polymer chains, 2) precision self-assembly of side chains, and 3) dynamic self-sorting and selective recognition. Typically, random copolymers bearing hydrophilic poly(ethylene glycol) and hydrophobic alkyl groups folded into small unimer micelles (~10 nm) via the association of the hydrophobic groups in water. Importantly, those random copolymers afforded precision intermolecular self-assembly into multichain micelles; the size, aggregation number, and thermoresponsive properties can be controlled as desired by tuning their molecular weight, composition, and side chains. The binary mixture of different random copolymers further self-sorted via chain exchange in water to simultaneously form discrete micelles. Namely, amphiphilic random copolymers can dynamically recognize themselves in complex media like natural biomolecules and proteins. Amphiphilic random copolymers opened new ways to create self-assembled materials with well-defined nanostructures and compartments, dynamic recognition properties, and functions.
Self-assembly of polymers and molecules is one of the promising strategies to produce functional materials with well-defined nanostructures in aqueous/organic solutions or in the solid/film state 1) , 2) , 3) , 4) , 5) , 6) , 7) . In water, amphiphilic copolymers comprising hydrophilic and hydrophobic segments self-assemble via the association of the hydrophobic segments to form micelles, vesicles, related aggregates, and hydrogels 1) , 2) , 3) , 4) , 5) , 6) . Those self-assemblies are applied to various research fields as nanoparticles, nanocapsules, delivery vessels, biomaterials, among many others 4) . To design self-assembled materials with desired properties and functions, controlling the size, three-dimensional architectures, and dynamic properties is essential. Self-assembly behavior and structures depend on the primary structure of amphiphilic copolymers: e.g., molecular weight (chain length), composition (hydrophilic/hydrophobic balance), monomer sequence distribution (block, gradient, random, alternating) 8) , 9) , 10) , 11) , 12) . Typically, amphiphilic block copolymers consisting of hydrophilic and hydrophobic chains induce intermolecular self-assembly of the polymer chains in water to form micelles whose hydrophobic cores are stabilized by the shell layer of multiple hydrophilic chains (Fig. 1b). In contrast, amphiphilic random copolymers bearing hydrophilic and hydrophobic side chains tend to fold via the self-assembly of the hydrophobic pendants in water (Fig. 1a). Thus, resulting unimer or multichain micelles turn smaller than micelles obtained from corresponding block copolymers. In fact, amphiphilic random or alternating copolyelectrolytes bearing hydrophobic alkyl pendants are known to fold into small micelles in water 10) , 11) , 12) . Such chain-folding process of synthetic polymers is also related to protein folding in biological systems 13) , 14) , 15) , 16) , 17) and thus intriguing as a strategy to create self-assembled materials with well-defined nanostructures.

Self-assembly of amphiphilic (a) random or (b) block copolymers into micelles in water.
Focusing on chain-folding properties and pendant self-assembly, we have recently developed controlled self-assembly systems of amphiphilic random copolymers bearing hydrophilic poly(ethylene glycol) and hydrophobic or functional pendants (Fig. 2) 14) , 18) , 19) , 20) . The self-assembly systems provide functional materials with well-defined nanostructures in aqueous and organic media: folded micelles, reverse micelles, vesicles, related aggregates, and hydrogels. For this, we prepared well-controlled random copolymers via living radical polymerization 21) . Typically, those random copolymers induced self-folding or intermolecular self-assembly into small micelles in water. The size, aggregation number, and thermoresponsive properties can be precisely controlled as desired by the primary structure (molecular weight, composition, pendant structures). Those copolymers further showed selective self-assembly and dynamic self-sorting (self-recognition) behavior via chain exchange process in complex aqueous media in the presence of other polymers. Multicompartment materials and sub 10-nm lamellar structure materials were also obtained with random copolymers by designing their pendants and monomer sequences. Herein, we report recent advances on the self-assembly systems of amphiphilic random copolymers for well-defined nanomaterials.

Self-assembly systems of amphiphilic random copolymers. (a) Self-folding of random copolymers bearing hydrophilic poly(ethylene glycol) (PEG) and hydrophobic, hydrogen-bonding, and fluorous side chains into unimer micelles in water. (b) Precision intermolecular self-assembly of random copolymers bearing PEG and dodecyl groups into size-controlled and thermoresponsive micelles in water. The size and aggregation number of the micelles can be controlled by tuning the composition and degree of polymerization (DP) of the copolymers. (c) Dynamic self-sorting of amphiphilic random copolymers into discrete micelles via chain exchange in water and self-healing and selectively adhesive hydrogels. (d) Pendant self-assembly of random copolymers for nanomaterials and microphase separation. (e) Functions of self-assembled polymers: catalysis, encapsulation, and bioapplications.
Amphiphilic random copolymers bearing hydrophilic PEG and hydrophobic alkyl pendants were designed for folded micelles in water 22) , 23) , 24) , 25) , 26) , 27) , 28) , 29) . Methacrylate-based random copolymers with controlled molecular weight and narrow molecular weight distribution (M w/M n<1.2) were efficiently synthesized by living radical copolymerization of PEG methyl ether methacrylate (PEGMA, M n=475, average number of oxyethylene units=8.5) and alkyl methacrylates (RMA, e.g., dodecyl methacrylate: DMA, butyl methacrylate: BMA) with a ruthenium catalyst [Ru(Ind)Cl(PPh3)2/n-Bu3N] and a chloride initiator (ethyl 2-chloro-2-phenylacetate: ECPA) in toluene at 80°C (Fig. 3) 22) , 25) , 26) , 27) . The degree of polymerization (DP) of the copolymers can be controlled by tuning the feed ratio of the monomers to the initiator: DP=40 - 600. PEGMA and RMA were simultaneously consumed at the same speed, independent of the alkyl chain length (R: -CH3, -C4H9, -C12H25, -C18H37 etc.) and the feed ratio of the two monomers (PEGMARMA=8/2 - 4/6, mol/mol). This indicates that the products are random copolymers without biased sequence distribution along a chain like gradient copolymers 9) . Acrylate or acrylamide-based random copolymers bearing PEG and dodecyl groups were also prepared by ruthenium or iron-catalyzed living radical copolymerization of their corresponding acrylates or acrylamides 28) , 29) . In contrast, living radical copolymerization of PEGMA and dodecyl acrylate (DA) gave an amphiphilic gradient copolymer whose DA content gradually increased from the initiating terminal to the growing terminal, because PEGMA (methacrylate) was faster consumed than DA (acrylate) in the copolymerization 28) . Thus, selection of monomer species is important to obtain random copolymers. Owing to high tolerance to polar functional groups, ruthenium-catalyzed living radical polymerization was also effective for the synthesis of amphiphilic random copolymers bearing hydrogen-bonding amide 30) and urea 31) groups or fluorous perfluoroalkyl groups 32) : PEGMA/BPUMA random copolymers 31) , PEGMA/13FOMA random copolymers 32) (Fig. 4).

Synthesis of amphiphilic random copolymers via living radical polymerization.
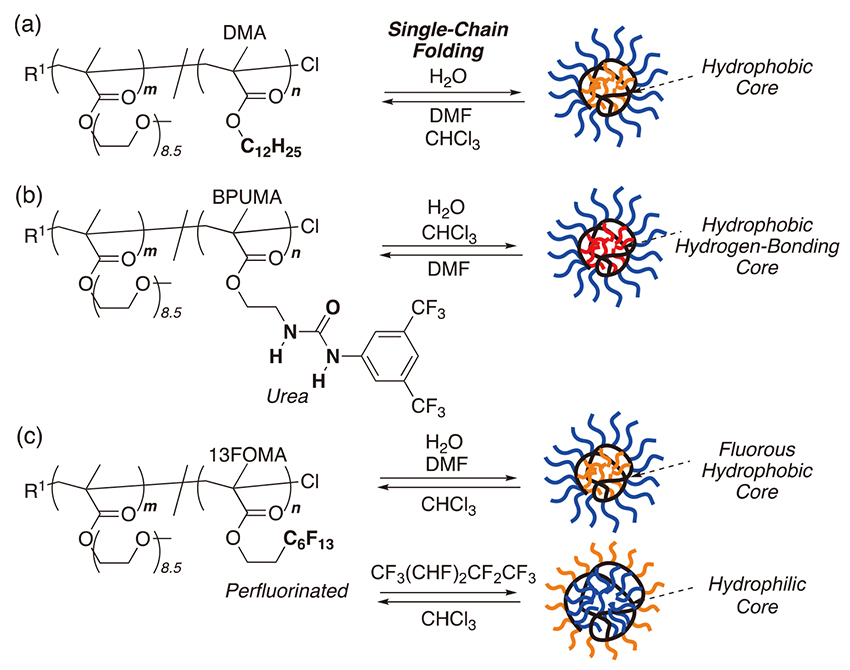
Self-folding of amphiphilic random copolymers into unimer micelles and reverse micelles in aqueous and organic media.
Self-folding of amphiphilic random copolymers into unimer micelles was examined in water or organic solvents (Fig. 4). The micellization and folded structures were analyzed by size-exclusion chromatography coupled with multiangle laser light scattering (SEC-MALLS), dynamic light scattering (DLS), nuclear magnetic resonance (NMR), transmission electron microscopy (TEM), and small angle X-ray scattering (SAXS). Here, PEGMA-based random copolymers with about 200 DP and 20 - 40 mol% hydrophobic or functional (hydrogen-bonding, fluorous) units were employed.
Typically, a PEGMA/DMA random copolymer (DP=200 DP, 40 mol% DMA) folded in water to efficiently form a compact unimer micelle (Fig. 4a) 22) , 25) . The absolute weight-average molecular weight of the copolymer in N,N-dimethylformamide (DMF, M w,DMF determined by SEC-MALLS) was in good agreement with M w,calcd of the single polymer chain that was calculated from the molecular weight determined by 1H NMR and the molecular weight distribution (M w,calcd=M n,NMR×M w/M n). Additionally, M w,H2O of the copolymer in water (determined by SEC-MALLS) was close to that in DMF (M w,DMF), while the size of the copolymer in water (Mp: peak-top molecular weight by SEC with poly(ethylene oxide) calibration, R h: hydrodynamic radius by DLS, R g: radius of gyration by SAXS) was smaller than that in DMF. This importantly demonstrates self-folding of the copolymer into a compact unimer micelle in water. The SAXS profile of the copolymer in water supported the formation of a globular micelle comprising a hydrophobic dodecyl core 25) . Folded random copolymer micelles were stable and maintained their sizes in water for a long time (>4 months) in a wide range of concentration (0.02 - 100 mg/mL) 25) , while the micelles were gradually unfolded by adding methanol into the aqueous solutions 22) .
Functionalization of amphiphilic random copolymers with hydrogen-bonding or fluorous pendants afforded self-folding not only in water but also in organic media. Random copolymers bearing PEG and hydrophobic urea pendants folded via the self-assembly of the urea groups in both water and chloroform (Fig. 4b) 31) . Amphiphilic and fluorous random copolymers bearing PEG and perfluoroalkyl pendants formed fluorous core micelles in water or DMF, while the copolymers folded into PEG-core reverse micelles in a fluorinated solvent [CF3(CHF)2CF2CF3] (Fig. 4c) 32) . Thus, folding of random copolymers can be controlled by designing their side chains.
We further investigated the effects of DP (chain length) and composition (hydrophilic/hydrophobic balance) on the self-folding and self-assembly behavior of the copolymers in water (Fig. 5) 25) , 26) , 27) . For this, PEGMA/DMA random copolymers with different DP (40 - 600) and DMA composition (20 - 50 mol%) were prepared by living radical polymerization 25) . The copolymers and their micelles were analyzed by SEC-MALLS in DMF and H2O. As a result, we found that the copolymers induced precision intermolecular self-assembly into multichain micelles with innovative controllability of size and aggregation number as follows 25) .

Precision self-assembly of PEGMA/DMA random copolymers in water: (a) Intermolecular self-assembly or self-folding in water; (b, c) absolute weight-average molecular weight (M w) of the copolymers or their micelles in DMF or water as a function of the chain length (DP: degree of polymerization, DMA= (b) 40 or (c) 50 mol%) 25) .
1) The copolymers have a clear threshold DP (DPth) between intermolecular self-assembly into multichain micelles and self-folding into unimer micelles. DPth increased with increasing DMA content in the copolymers.
2) The copolymers shorter than DPth exclusively induce intermolecular self-assembly in water. The resulting multichain micelles have uniform and constant size, independent of DP (Fig. 5b, c). The size of their micelles was identical to that of a unimer micelle obtained from a DPth copolymer.
3) The size of micelles (<DPth) increased with increasing the hydrophobic DMA content: e.g., 40 mol% DMA copolymer micelles (DP<200) : M w,H2O=~100000, 50 mol% DMA copolymer micelles (DP<250) : M w,H2O=~220000, (Fig. 5b, c).
4) If DP of the copolymers is shorter than DPth, aggregation number of multichain micelles (N agg=M w,H2O/M w,DMF) can be controlled by tuning DP and composition. In the case of 40 mol% DMA copolymer micelles: N agg (DP) = 1 (190), 2 (102), 3 (68), 4 (52), and 5 (44). This means that N agg of micelles is also controlled by living radical polymerization.
5) Copolymers with broad molecular weight distribution (<DPth, synthesized by free radical copolymerization) also form uniform micelles in water. The size is identical to that of micelles obtained from well-controlled random copolymers (prepared by living radical copolymerization), because polymer chains close to DPth self-fold and those shorter than DPth intermolecularly self-assemble.
Such unique self-assembly in water universally occurred for amphiphilic random copolymers with different alkyl groups 26) or main chains (acrylate 28) , acrylamide 29) ). Owing to thermoresponsive PEG units, their random copolymer micelles showed lower critical solution temperature (LCST)-type solubility in water 22) , 23) , 24) , 25) , 26) , 27) , 28) , 29) , 30) , 31) , 32) . PEGMA/RMA random copolymers afford dual control of both size and cloud point temperature of their micelles by tuning their alkyl groups (R) and RMA composition (Fig. 6) 26) . The size of their micelles (M w,H2O) is determined by the weight fraction of the hydrophobic segments (HWF) of their copolymers. Random copolymers comprising 60 mol% BMA (-C4H9) or 50 mol% OcMA (-C8H17) or 40 mol% DMA (-C12H25) had close HWF values (42 - 45 wt%) to form micelles with almost identical size (M w,H2O=~100000), while their cloud point (Cp) temperatures were different. In contrast, random copolymers comprising 50 mol% RMA of different alkyl groups formed different size micelles with identical Cp temperatures; the size increased with increasing the length of the alkyl groups. Thus, PEGMA/RMA random copolymers provide thermoresponsive and size-controlled micelles in water. The size and thermoresponsive properties of random copolymer micelles can be also tuned by the PEG length 27) .

Size-controlled and thermoresponsive micelles of PEGMA/RMA random copolymers in water 26) .
PEGMA/RMA random copolymers induce self-sorting (selective self-assembly; self-recognition) in water in the presence of other copolymers comprising different alkyl pendants and composition (Fig. 2c) 25) , 26) , 28) , 33) . For example, if 50 mol% DMA random copolymers were mixed with 30 mol% DMA random copolymers in water, their copolymers recognize themselves and find identical polymer pairs in water to simultaneously form discrete multichain micelles with different sizes [M w,H2O=~220000 (50 mol% DMA), ~50000 (30 mol% DMA)] 25) , 33) . Similarly, the binary mixture of 50 mol% DMA random copolymers and 70 mol% BMA random copolymers also self-sorted to discrete multichain micelles comprising dodecyl cores or butyl cores though their micelles have almost identical size in water (M w,H2O=~220000) 33) . Interestingly, their copolymers dynamically induce chain exchange between identical micelles in the presence of other micelles (Fig. 2c) 33) . Such self-sorting systems have been generally realized by using elaborated molecules such as supramolecular compounds 34) , 35) , 36) , 37) , 38) and peptide derivatives 39) , 40) . Thus, self-sorting of simple random copolymers is quite unique.
Owing to dynamic self-assembly and self-sorting behavior, ABA triblock copolymers comprising PEGMA/RMA random copolymer A segments and a hydrophilic PEG B segment gave self-healing and selectively adhesive hydrogels (Fig. 7) 33) , 41) . The cut B70-PEG-B70 hydrogels (B70: PEGMA/70 mol% BMA random) were contacted with each other at 25℃ for 2 hours to be healed via dynamic chain exchange. In contrast, B70-PEG-B70 hydrogels were never adhered with D50-PEG-D50 hydrogels (D50: PEGMA/50 mol% DMA random) due to the selective chain association (self-sorting) on the gel surfaces.

(a) Self-healing and (b) selectively adhesive hydrogels of ABA triblock copolymers comprising amphiphilic PEGMA/BMA (70 mol%, B70) or PEGMA/DMA (50 mol%, D50) random copolymers (A) and a hydrophilic PEG (B) 33) .
Self-folded micelles, single-chain polymer nanoparticle, and related nanoaggregates obtained from amphiphilic random copolymers are useful as nanoreactors 30) , 42) , 43) , nanocapsules 22) , 23) , 24) , 25) , 44) , 45) , and biomaterials 46) , 47) . Self-folded micelles and single-chain polymer nanoparticles work as polymer catalysts containing unique nanospaces and cavities like proteins. For example, a folded polymer iron catalyst efficiently induced living radical polymerization of various methacrylates including methacrylic acid to show high activity and tolerance to functional groups (Fig. 8a) 43) . Easy product recovery and catalyst recycle were also achieved. The polymer catalyst was prepared by the intramolecular crosslinking of a self-folded amphiphilic random copolymer bearing hydrophilic PEG, hydrophobic dodecyl groups, and urea-functionalized aniline units with 2,6-pyridinedicarboxaldehyde. The resulting bis (imino) pyridine spacers play a dual role of crosslinking units and ligand cavities for the complexation to irons. Amphiphilic fluorous random copolymers reversibly form fluorous core micelles and aggregates in water and PEG core counterparts in a fluorinated solvent 32) . The former fluorous core aggregates were useful as polymer materials for protein conjugation (Fig. 8b) 46) . The latter PEG core aggregates were effective for polymer nanocapsules that stably preserve proteins in a fluorinated solvent; the proteins recovered from the capsules still maintained enzymatic activity (Fig. 8c) 47) . Thus, amphiphilic fluorous random copolymers are new candidates as polymer biomaterials different from hydrophilic polymers generally utilized for such applications.
Amphiphilic random copolymers form not only folded micelles but also various nanostructure materials including double core polymers and micelles 48) , 49) , 50) , multicompartment micelles 48) , 49) , 50) , necklace micelles 29) , 50) , flower micelles 51) , and vesicles 52) in water (Fig. 2d). For example, A/C-B/C amphiphilic random block copolymers comprising hydrophilic PEG (A) and different hydrophobic pendants (B: dodecyl, C: benzyl) induced double folding via the orthogonal self-assembly of their hydrophobic pendants in water; the intramolecular crosslinking of the double core micelles selectively gave double core single-chain polymers 48) . An amphiphilic tadpole polymer consisting of an amphiphilic crosslinked polymer nanoparticle and a hydrophobic polymer chain formed multicompartment micelles in water 48) . Amphiphilic/fluorous random block copolymers were also effective for such double core and multicompartment micelles 49) . Recently, we developed selective coupling systems of amphiphilic folded polymer micelles via physical interaction in water for double core micelles, alternating necklace micelles, and hydrogels 50) . Those micelle-based self-assemblies afford dynamic structure change and cleavage of the micelle nanodomains by tuning solvents and pH and adding salts.
Introduction of crystalline side chains into amphiphilic polymers is also a promising strategy to produce well-defined nanostructure materials 51) , 52) . Typically, random copolyacrylates carrying PEG and octadecyl groups induced microphase separation of the pendants via the crystallization of the octadecyl groups to form lamellar structures with 5 - 6 nm domain spacing in the solid state 52) . The hydration of the thin films led to the formation of polymer vesicles in water.
We demonstrated self-assembly systems of amphiphilic random copolymers to produce folded micelles, vesicles, related aggregates, hydrogels, and sub-10 nm microphase separation materials. In contrast to that of block copolymers, self-assembly of those random copolymers is driven by the association of the side chains and the self-folding of the main chains. As a result, this technique is quite effective to build small sub-10 nm structures and domains into polymer materials like proteins and living organisms, though fine control of sub-10 nm scale self-assembly was generally difficult so far by using block and related copolymers. Therefore, amphiphilic random copolymers would open new vistas to create self-assembled nanostructure materials with well-defined architectures, desired properties, and on-demand functions for various applications and research fields.
This work was supported by Japan Society for the Promotion of Science KAKENHI Grants (JP17H03066, JP17K19159, JP19K22218), by The Ogasawara Foundation for the Promotion of Science & Engineering, by The Noguchi Institute, by Inamori Foundation, and by Tokuyama Science Foundation.
