Review
Inner and Interfacial Environmental Nanoarchitectonics of Supramolecular Assemblies Formed by Amphiphiles: from Emergence to Application

2023 Volume 72 Issue 2 Pages 105-116
Details
2023 Volume 72 Issue 2 Pages 105-116
The inner and interfacial environments of self-assemblies provide fascinating nano-space for selective and efficient chemical reactions and processes. In biological systems, various chemical reactions, molecular recognition, and transport occur precisely and selectively by virtue of effective molecular interactions on biological membranes and proteins. Considering these advantages and the concept of nanoarchitectonics, we demonstrated that the photochromism of a lophine dimer was accelerated by using confined nano-spaces formed by surfactant micelles. The photoresponsive micelles were used for the rapid controlled release of a model drug upon ultraviolet light irradiation. Furthermore, selective ion recognition inside the self-assembled molecular films at the interfaces was investigated. The anion-π interaction between the anion and an electron-deficient aromatic ring was evaluated on a solid substrate modified with a naphthalenediimide (NDI) analog. Force curve measurements afforded a quantitative analysis of anion-π interactions on the NDI film. The strength of anion-π interactions is regulated by the electric fields on the electrode. An optical probe was developed to visualize the distribution of Cs ions in the soil, plant bodies, and aqueous media using an optode system. Advances in the development of molecular functional systems are expected based not only on molecular structures but also on the spaces and environments produced by them.
In biological systems, various chemical reactions, molecular recognition, and transport occur precisely and selectively by virtue of effective molecular interactions on biological membranes and proteins. These structured nano-spaces provide recognition sites for specific host-guest interactions. For example, enzymatic reactions occur in specific pockets formed by proteins and assemblies. When certain multifunctional proteins, e.g. cytochrome c, are embedded in biological membranes, the enzymatic activity and membrane interactions are strongly affected 1) . In recent years, artificial enzymes have been produced from synthetic functional proteins that realize “molecular evolution” 2) . Artificial molecular spaces have also been studied in the field of supramolecular chemistry, and are dedicated to applications such as molecular recognition, sensing, and catalysis with the supramolecular assemblies and complexes 3) . Fujita et al. reported that the [2+2] cross-photodimerization of olefins proceeds efficiently and selectively in a self-assembled coordination cage 4) . This shows that the confined nano-space affects the reaction pathway and collision frequency of the substrates 5) , 6) . Kunitake and Ariga found that the hydrogen bond between the phosphate and guanidino groups was remarkably enhanced at water interfaces. In particular, hydrogen bonds are enhanced on a Langmuir monolayer at the air/water interface 106-7-fold in comparison with those in the bulk aqueous medium owing to the low permeability of interfacial water 7) . This report indicates that molecular assemblies at the interfaces (particularly at the air/water interface) provide ideal spaces for molecular recognition.
These reports indicate that the interior and interface of molecular assemblies provide a fascinating space for chemical processes. Interesting features of the inner and interfacial environments of the molecular assemblies are shown in Figs. 1 (a) -1 (c). (a) The “confined” space enhances specific chemical reactions owing to host-guest chemistry and the concentration effect of these substrates. (b) Environments with different polarities (e.g., polar and nonpolar media) can coexist in a medium. For example, nonpolar spaces can be produced in aqueous surfactant solutions owing to micelle formation. (c) Because nano-spaces formed by molecular assemblies are soft and flexible, their size and morphology, which are crucial for the recognition and inclusion of substances, can vary with external stimuli, which enables the controlled release of drugs and active components.

Examples of previous and this works of functional systems by using the inner and interfacial environments of molecular assemblies.
Recently, new nanotechnology with self-assembly of functional molecules is defined as “nanoarchitectonics” and achieved novel functional systems across scales 8) . This review highlights the recent progress in our research on the development of supramolecular functions based on nanoarchitectonics using the inner and interfacial environments of self-assemblies, formed by the amphiphiles: (1) photo-induced rapid controlled release using the inner environments of molecular assemblies (Fig. 1 (d)) and (2) ion recognition inside self-assembled molecular films at solid/liquid interfaces (Fig. 1 (e)).
Amphiphilic compounds and surfactants form a variety of molecular assemblies, such as micelles and vesicles, in water. Aqueous solutions of molecular assemblies can solubilize water-insoluble or poorly water-soluble compounds when incorporated into molecular assemblies. This solubilization technique is used to dissolve active substances in aqueous media and is an indispensable step in the production of commercial beverages, foods, cosmetics, medicines, and household products.
The controlled assembly of amphiphilic compounds causing deformation and/or morphological changes can be applied to regulate the release of drugs and other active compounds (Fig. 2 (a)). To trigger morphological changes in molecular assemblies, external stimuli are applied, such as light, redox reactions, magnetic fields, and temperature or pH changes 9) , 10) . Among these, light is a promising external stimulus because it is clean and remotely applied with a high spatial resolution and specific wavelength. Therefore, many photoresponsive amphiphilic compounds have been developed, and the formation of molecular assemblies, such as micelles and vesicles, by photoirradiation has already been realized 9) , 11) , 12) .

(a) Schematic image of controlled release with a photoresponsive molecular assembly. (b)Photochromism of the lophine dimer.
For decades, photoinduced morphological changes in molecular assemblies and their applications have been accomplished. For example, our group demonstrated the controlled release of volatile oils as model perfumes with an azobenzene-type cationic surfactant that exhibits photoswitchable formation of micelles upon ultraviolet (UV) irradiation 13) , 14) . However, to induce significant changes, these conventional systems require minutes to hours of photoirradiation. For practical applications, the response time needs to be fast, requiring a change in solution properties at an arbitrary moment that ceases when the stimulus stops (Fig. 2 (a)). Thus far, there have been no attempts to speed up the controlled formation of photoresponsive molecular assemblies.
To achieve rapid control of amphiphile self-assembly, we investigated photochromic compounds showing rapid changes in molecular structure and focused on the lophine dimer (LPD) as a photochromic moiety. The LPD molecules quickly dissociated into two lophyl radicals and were recovered by a thermal recombination reaction (Fig. 2 (b)) 15) . Although this recombination is extremely slow, it has been reported that the reaction rate is significantly enhanced by inhibiting free diffusion of the radical species. This was achieved by connecting two lophine moieties through a linker 16) , 17) , 18) or confining them to microscopic domains formed by ionic liquids 19) , 20) .
Our group demonstrated rapid photoisomerization of LPD using micelles formed by surfactants. Alkylated lophine dimers (LPD-C6 and C12) (Fig. 3) were solubilized in aqueous solutions of cetyltrimethylammonium bromide (CTAB). The recombination of lophyl radicals produced by UV light irradiation from the dimers was characterized by UV/vis absorption spectroscopy. For the alkylated LPD in the aqueous CTAB solutions, UV irradiation led to the appearance of a new absorption band at 619 nm, attributed to a lophyl radical. When the solution was kept in the dark, the absorption intensity of the band decreased, reaching a stationary state. The plots illustrate the inverse of the absorption at 619 nm (1/A) versus time (t). The linearity of these plots indicates that the recombination follows a second-order rate law not only in organic solvents 21) , but also in micelles. The apparent reaction rates (k) were determined from the slopes. The apparent recombination rates were 100-fold higher than those in octane, where the lophyl radicals diffused freely 21) . It is likely that the rate enhancement is derived from the inhibited diffusion of lophyl radicals in the micelle, which acts as a nano-cage. This result reveals that the inside of the micelles is suitable for inducing rapid structural changes in the LPD molecule.

Chemical structures of lophine dimer derivatives.
To further enhance recombination, micelles composed of amphiphilic lophine dimers were prepared to shorten the diffusion distance of the lophyl radicals. Amphiphilic lophine dimers bearing six or two triethylene glycol (TEG) groups (3TEG-LPD or TEG-LPD, respectively) were synthesized 22) . 3TEG-LPD formed micelles in water (critical micelle concentration, cmc: 0.80 μM). The water-insoluble TEG-LPD was solubilized in an aqueous CTAB solution.
The apparent reaction rates of the recombination of lophyl radicals (k’) are shown in Fig. 4. The k’ value of 3TEG-LPD in the micelles was 1.4 s−1, which was accelerated by ~800-fold compared to that in tetrahydrofuran (THF), where the micelles were not formed and the produced radicals diffused freely 22) . The rate was 100-fold higher than that reported for lipophilic C6-LPD in CTAB micelles. The recombination rate of the TEG-LPD/CTAB mixture in water was 50-fold higher than that in the organic solvent. The rate enhancement in the 3TEG-LPD micelle was better than that of TEG-LPD, as TEG-LPD was diluted in CTAB micelles, resulting in the free diffusion of lophyl radicals.
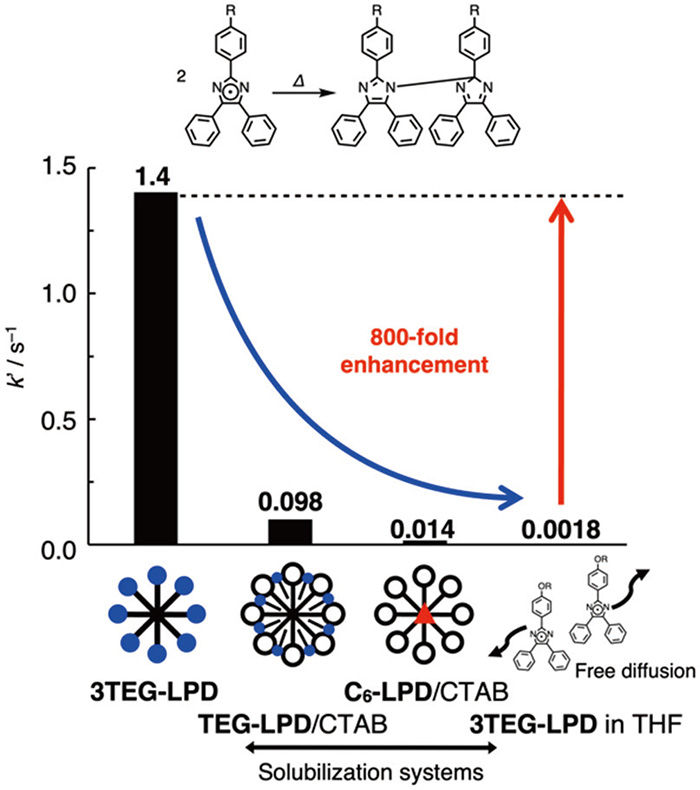
Apparent reaction rates of the recombination (k’/s−1) of the aqueous solutions of 0.5 mM 3TEG-LPD, 0.5 mM TEG-LPD/50 mM CTAB, and saturated C6-LPD/50 mM CTAB, and THF solution of TEG-LPD.
It was indicated that the concentration (number density) of LPD, governed by the number of LPD and volume of the reaction space, has a significant impact on the recombination rate 22) , 23) . To confirm this, TEG-LPD was solubilized in micelles formed by cationic surfactants with different alkyl chains (C12, C14, or C16). The recombination rate in the micellar solution of the surfactant with a shorter alkyl chain was enhanced compared to that of the surfactant with a longer alkyl chain 24) . Small-angle neutron scattering (SANS) measurements showed that mixtures of the cationic surfactant and lophine dimer derivative formed elliptical micelles, and a smaller inner volume was observed when shorter chain surfactants were used. Furthermore, the evaluation of the kinetic parameters indicated that the randomness of the lophyl radical in the shorter surfactant micelles was higher than that in the longer surfactant micelles. These results reveal that the size of the confined space formed by the micelles determines the recombination rate of the lophyl radicals.
2.2 Rapid morphological changes of the micelles formed by amphiphilic lophine dimers with photoirradiationRapid control of the static surface tension of the aqueous 3TEG-LPD solution by photoirradiation was examined. The constant surface tension value of 5.0 mM aqueous 3TEG-LPD solution was observed to be approximately 46.5 mN/m, in the dark. When the solution was irradiated with UV light, surface tension decreased within a few seconds. After a certain duration, the surface tension immediately recovers to its original value in the absence of photoirradiation. These reversible changes were observed repeatedly. The decrease in the surface tension value upon UV irradiation suggests that the lophyl radical produced, which is less bulky than the initial dimer form, efficiently adsorbs at the air/water interface. After the irradiation was halted, the recombination of the radical species readily proceeded in the micelles of the bulk phase or in the Gibbs monolayer formed at the air/water interface. Therefore, we achieved reversible and quick control of the surface tension using fast dissociation of the lophine dimer upon photoirradiation and accelerated the recombination of lophyl radicals in the micelles 22) .
Next, we examined the rapid morphological changes in 3TEG-LPD micelles upon UV irradiation. However, conventional particle-sizing equipment using light scattering and laser diffraction is problematic for detailed morphological analysis of molecular assemblies and their rapid changes under light irradiation. Accordingly, we developed an in situ SANS system by installing a mercury lamp and a UV/Vis absorption spectrometer to monitor the morphology of the micelles and the photochromic reaction of the LPD moiety 25) . Figure 5 (a) shows the SANS profiles of 3TEG-LPD in D2O. The experimental data were well fitted using a uniform prolate ellipsoid model with a long radius (r a) of ~47 Å and a short radius (r b) of ~28 Å. UV irradiation caused an increase in the scattering intensity toward the lower q region, which reached a constant state within 2 min. The post-irradiation SANS profile was also well fitted using a uniform prolate ellipsoid model. The short radius remained at ~28 Å, and the long radius increased to ~70 Å, indicating that UV irradiation induced elongation of the longer axis of the micelles. This is because the hydrophobic portion of the resulting lophyl radical has a relatively larger volume than that of the original dimer form, whose LPD unit is tightly linked with a covalent bond, and photoirradiation causes the formation of micelles with a lower curvature.

Results of simultaneous in-situ SANS and UV/Vis absorption. (a) SANS profiles of 10 mM 3TEG- LPD in D2O before and after 2 min UV irradiation and 4 min standing in the dark with curve fitting. (b) The integrated scattering intensity of the SANS profiles of 3TEG-LPD in the q-region of 0.01-0.05 Å−1 during cycles of irradiation and standing in the dark. (c) UV/Vis absorption spectra before and after UV irradiation and standing in the dark (right), and the temporal changes in absorption at 580 nm during the cycle using an in-situ UV/Vis absorption spectrometer (left).
When UV irradiation ceased, the SANS profile readily recovered to its initial state (Fig. 5 (a)). Figure 5 (b) shows the integrated scattering intensity in the q-region of 0.01-0.05 Å−1, for the scattering profiles at 60 s intervals during cycles of 2 min UV irradiation followed by 4 min standing in the dark. This observation revealed that the elongation of the elliptical micelles was completed after 60 s of irradiation and reversed after 60 s of standing in the dark. These reversible changes in the SANS profiles were repeatable over ten cycles. UV/Vis absorption spectra were collected simultaneously during the SANS measurements. As shown in Fig. 5 (c), a characteristic absorption band of the lophyl radical was observed at 580 nm during UV irradiation. The temporal change in the absorption at 580 nm during the irradiation cycle revealed that morphological changes in the micelles and production of lophyl radicals occurred without a time lag within the analytical time resolution (Figs. 5 (b) and 5 (c)). These results indicate that the reorganization of the surfactant molecules in the micelles following photoisomerization was complete within the timescale of the SANS measurements.
Interestingly, the photoisomerization and morphological changes in the molecular assembly of amphiphilic lophine dimers readily proceed even in highly concentrated systems, unlike azobenzene derivatives, which are the most popular photochromic compounds. Azobenzene derivatives show a lower rate and yield of photoisomerization in a confined system 26) or in the solid state 27) because sufficient free volume is necessary for conformational changes in the molecular structure during trans-cis isomerization. Therefore, the lophine dimer analog is suitable for photoresponsive functional systems based on molecular assemblies or supramolecules.
2.3 Rapid controlled release of a model drug with photoirradiationThese results show that UV irradiation induced rapid and reversible changes in the morphology of the 3TEG-LPD micelles. Using this promising photoresponsive surfactant, substances solubilized in micelles could be readily released by UV irradiation 25) , 28) . To demonstrate the controlled release, we prepared an aqueous solution of 3TEG-LPD containing calcein as a fluorescent model drug. For a 5.0 mM 3TEG-LPD solution with calcein at the solubilization limit (1.0 mM) under UV irradiation, the band at 545 nm assigned to calcein in the micelles decreased, with the peak shifting to 556 nm assigned to aggregated calcein molecules in water. This indicates that calcein molecules were incorporated into micelles released under UV irradiation. Figure 6 shows temporal changes in the relative intensities of the fluorescence peaks. The intensity reached a constant state within 60 s of UV irradiation, which is slightly shorter than the change in the SANS profile. We assumed that UV irradiation for 60 s induced morphological changes in the micelles, affecting the solubilization state of calcein. In contrast, the fluorescence spectra of the calcein/3TEG-LPD aqueous solution remained unchanged in the absence of UV irradiation (Fig. 6), indicating that UV light induced the release of calcein from the inside of the 3TEG-LPD. The fluorescence of calcein in the 6TEG-LPD micelles remained unchanged upon UV irradiation (Fig. 6), corresponding to the nonresponsive behavior of 6TEG-LPD. These results revealed that the morphological change in the 3TEG-LPD micelles under UV irradiation caused the rapid and effective release of calcein. This rapidly controlled release system allows for effective and on-demand delivery of active components, such as drugs and perfumes.

Normalized transient changes in fluorescence intensity at the peaks for 1.0 mM calcein/5.0 mM 3TEG-LPD in the absence and presence of UV irradiation and 6TEG-LPD aqueous solutions in the presence of UV irradiation. The excitation wavelength is 365 nm.
Interfacial phenomena, such as wettability, friction, adhesiveness, and dispersion of colloidal particles, are important factors in determining the material characteristics. The interfacial properties are mainly governed by physicochemical properties, such as charge, hydrophilic-lipophilic balance, and surface morphology. Adsorption at solid/liquid interfaces based on intermolecular interactions has a significant impact on the interfacial properties. For example, amphiphilic molecules reversibly adsorb at a solid/liquid interface via electrostatic or hydrophobic interactions and contribute to the control of friction and colloidal dispersibility.
Anion-π interactions are noncovalent interactions between an electron-deficient aromatic ring and anion species 29) , 30) . When a strong electron-withdrawing group is introduced into an aromatic system, the charge distribution on the aromatic plane is inverted, and the negative quadrupole moment (Q zz) attracts anions 30) . Naphthalenediimide (NDI), bearing a Q zz of 18 B, is an excellent acceptor for anion-π interactions. Conversely, electron-rich aromatic compounds such as benzene attract cation species via comparable cation-π interactions 31) . Although anion-π interactions have been proposed by theoretical investigations, they have recently been observed in crystal structures and solutions 30) , 32) , 33) , 34) , 35) , and have contributed to functional molecular systems 36) , 37) , 38) . Matile et al. reported the transport of anions across biological membranes 39) and chemo- or enantioselective catalysis 40) . Simultaneously, it was revealed that many protein-folding structures and enzymatic reactions are supported by anion-π interactions 41) , 42) . Furthermore, Zeng et al. revealed that anion-π and cation-π interactions coexist during bioadhesion of catecholic moieties with phosphate ester and potassium 43) . These reports reveal that anion-π interactions are excellent tools for controlling not only small molecular functions but also macromolecular and macroscale phenomena at material interfaces.
The nanomechanics of molecular interactions have been examined by force measurements using surface force apparatus (SFA) or atomic force microscopy (AFM). Israelachvili evaluated cation-π interactions in the adhesion proteins of marine organisms in aqueous media using force analysis and observed that these interactions play a key role in the underwater adhesion of marine creatures, such as mussels 44) . As mentioned above, anion-π interactions also contribute to the biological adhesion process (SFA measurements) 43) . Inhibition experiments with these anions revealed that the strength of anion-π interactions was related to the order HPO4 2−>SO4 2−>NO3 −. This trend is associated with the charge density, polarity, and hydration behavior of the anions. This report demonstrated that interfacial analytical techniques have revealed anion-π interactions at solid/water interfaces and their contributions to interfacial phenomena. This is the only report of anion-π interactions at the interface. However, this system has been investigated using complex biological molecules. The system must be simplified to understand the interactions at the interface.
To examine the anion-π interactions at the solid/liquid interface, we immobilized an NDI derivative (TEG-NDI-TES) composed of an NDI core as a popular and simple anion-π acceptor and an alkoxysilyl group capable of covalent bonding with the silica surface (Fig. 7 (a)). Based on anion-π interactions, the anion species were adsorbed on NDI@silica. For quantitative evaluation, the adsorption amounts of the anion species were measured using a quartz crystal microbalance with dissipation (QCM-D), which assumes the adsorbed mass (Sauerbrey mass, Δm [ng]) owing to changes in the frequency of the QCM-D sensor (ΔF n [Hz]) using the following equation: Δm=−C× (ΔF n/n), where C is a constant (0.177 mg/m2 • Hz) and n is the overtone number (3 in this study). Pure water was flowed onto the QCM-D silica sensor, modified with TEG-NDI-TES, and then an aqueous solution of tetrabutylammonium chloride (TBACl) was continuously injected, which resulted in a decrease in ΔF 3, as shown in Fig. 7(b).

(a) Chemical structure of TEG-NDI-TES and immobilization on the silica substrate. (b) Variation in the frequency (ΔF 3/3) as a function of time. TBACl concentration was fixed at 1.0×10−5 M. (b) Plots of the adsorption amount of the anions (Γ anion) vs. the hydration energy of anion species (ΔG anion).
We also examined the adsorption behavior of NDI@silica for several anions (Fig. 7c). The strength of anion-π interactions depends on the electron density of the anions 39) , which corresponds to their hydration energy (ΔG hyd). As shown in Fig. 7c, the plots of Γ anion vs. ΔG hyd revealed that halide anions (Cl−>Br−>I−) and SO4 2−, and the anions with larger ΔG hyd showed larger adsorption amounts. However, NO3 − specifically showed a larger Γ anion. A previous report presented similar results in that anions with a π-conjugated system show better anion binding with the NDI units than halide anions because of the concerted effect of π-π interactions with anion-π interactions 39) . From these results, we observed the trend of anion adsorption on the NDI-modified substrate, which follows the strength of anion-π interactions.
Next, to investigate the anion-π interactions at the solid/liquid interface, we recorded force curve measurements using atomic force microscopy (AFM), which can measure the physical forces between NDI@silica and a silicon nitride cantilever in aqueous media. The surface of the cantilever is negatively charged in water under neutral conditions because the pK a of silicon nitride is ~5.5. Figure 8a shows a typical force curve of NDI@silica with a negatively charged cantilever in pure water. From the apparent separation of 3-4 nm, an attractive force of ~0.1 nN was observed. The curve was different from the theoretical curve of the van der Waals attractive force under the assumption of a general organic thin film. In contrast, the ethylene glycol-modified silica substrate (TEG@silica) showed a repulsive force with respect to the molecular length, indicating that the attractive force was derived from the NDI moiety (Fig. 8b). The TEG groups on NDI@silica also exhibited a similar repulsive force according to the steric effect.

Approaching force curves of NDI@silica in pure water (a), TEG@silica substrate in pure water (b), and NDI@silica in 10 mM TBACl of aqueous solution (c). The theoretical curve of van der Waals force is attached on the curve of NDI@silica in pure water. The Z-scale interval is 0.2 nN.
Furthermore, to clarify the anion-π interactions at the interface, we examined the effect of chloride anions. According to the Derjaguin-Landau-Verwey-Overbeek (DLVO) theory, the repulsive force is suppressed in the presence of salts owing to the electrostatic shielding on the interfaces and the net attractive force in the presence of salts and can be enhanced compared to that in the absence. However, if the attractive force is derived from the interaction between NDI@silica and the cantilever surface, the excess amount of added chloride anions are likely to interact with the NDI moieties on the surface and inhibit the attractive force, resulting in a repulsive force owing to electrostatic repulsion. As a result, a repulsive force was observed, which then jumped into an attractive force in the presence of 10 mM TBACl. This revealed that the electrostatic repulsive force was canceled when the negatively charged cantilever entered the NDI film (Fig. 8c). To observe the clear trend of this inhabitation behavior of the added chloride anions, a histogram of the detected force was created, which confirmed that the attractive force gradually reverted to the repulsive force with increasing chloride ion concentration. These results revealed that anion-π interactions were observed on NDI@silica. The effect of anion species on inhibition was examined using 10 mM TBACl, TBABr, and TBAI in water. According to these histograms, the inhibition behavior trend was Cl−>Br−>I−, which corresponds to the trend of the adsorption amount obtained by the QCM-D measurements and a previous report 39) . The results of this study show that the stronger the anion-π interaction between NDI@silica and the species, the greater the amount of anion adsorption, resulting in a higher probability of repulsive force. Finally, using an autocorrelation function analysis 45) , we assumed a single-molecule force of anion-π interactions at the interface. The autocorrelation coefficient, according to the detailed histogram of the attractive force observed on NDI@silica in pure water, revealed a peak periodicity with an interval of ~40 pN. This represents the single-molecule force of the anion-π interactions observed in this study. The single-molecule anion-π interactions are comparable to those of other molecular interactions, such as cation-π interactions with calixarene-ammonium cations (~80 pN) 46) and the strong antigen-antibody interaction with avidin-biotin (160 pN) 43) , suggesting that the obtained value of the single-molecule force is sufficient for the interaction. To the best of our knowledge, this is the first example of a single-molecule force for anion-π interactions 47) .
3.2 Control of anion-π interactions at solid/water interfaces using an electric fieldTo elaborate on remote control with electric fields, an organocatalyst working via anion-π interactions was immobilized on conducting surfaces. The application of an electric field is expected to polarize the π-acidic aromatic system and convert Q zz>+10 B into an induced dipole μ z (Fig. 9 (a)). The tightened binding of anionic intermediates and transition states on this polarized π surface is reflected in the increased catalytic activity. To elaborate on these expectations, we designed, synthesized, and evaluated a heterogeneous anion-π catalyst (NDI@ITO), inspired by sophisticated anion-π catalysts 36) , 48) (Fig. 9 (c)). The proposed bifunctional motif combines the privileged π-acidic surface provided by naphthalenediimides (NDIs, Q zz~+18 B) with a tertiary amine. Interfacing with a conformationally constrained Leonard turn has been shown to be perfect for reactions on aromatic surfaces.

(a) Enhancement of the anion-π interaction and a catalytic reaction by induced polarization of the π electron cloud under an electric field on an ITO electrode. (b) With malonic acid half thioester (MAHT), enolate addition to yield a product (A) and decarboxylation to a product (D) are in kinetic competition. (c) Chemical structure of a catalyst bearing phosphonic acid moieties. (d) Dependence of A/D product ratio for the potential applied to the catalyst on the electrode (NDI@ITO).
To immobilize this bifunctional catalyst on the ITO surfaces, diphosphonate feet were introduced via sulfide substituents in the NDI core. For the electric-field-assisted catalysis, the heterogeneous catalyst was immersed in THF containing 200 mM MAHT and 2 M trans-β-nitrostyrene. Hexafluorophosphate (PF6) salts were used as the electrolyte (tetrabutylammonium: TBAPF6, 0.1 M) and a reference electrode (Ag/AgPF6) to minimize interference from competing anion-π interactions on the catalyst. The ratios of addition product A and decarboxylation product D, that is, the A/D selectivity, were determined by 1H-NMR spectroscopy (Fig. 9 (d)). At 0 V against Ag/Ag+, A/D=0.080 was obtained, indicating that decarboxylation of product D clearly dominates under these conditions. The application of increasingly positive potentials caused an almost linear increase in A/D selectivity until saturation was reached around A/D=1.8 (Fig. 9d). This behavior corresponds well with the theoretical predictions of the dependence of anion-π interactions on the electric field 49) , 50) , 51) .
The inversion of selectivity originates from the selective acceleration of the intrinsically disfavored but relevant enolate addition reaction toward product A. A comparison of initial rates at 0.0 V and +0.5 V revealed that the application of an electric field to anion-π catalyst results in a rate enhancement of v/v 0=190 for the formation of addition product A. In contrast, a nearly negligible rate enhancement (v/v 0=1.3) was found for the formation of decarboxylation product D on the anion-π catalyst in electric fields. The different rate enhancements calculated to transition state stabilization of ΔE a=−15.5 kJ/mol for addition and ΔE a=−0.7 kJ/mol for decarboxylation. Thus, the effect of electric fields to selectivity of transition-state recognition amounted to ΔΔE a=−14.8 kJ/mol. This coinciding enhancement of the rate and selectivity is in agreement with the fundamental principles of catalysis. Moreover, according to our earlier findings using NDI catalysts with varying π-acidity 52) , the selective acceleration of enolate addition was consistent with enhanced anion-π interactions, which were caused by polarization of the π surface in the catalyst by electric fields. Although convincing and consistent, this interpretation does not exclude other explanations for the identified remote control of anion-π catalysts in electric fields.
3.3 Cesium recognition and visualization at the interfacesIn recent years, methods for monitoring, detecting, and removing invisible environmental hazards have received attention for safe and healthy surroundings. Radioactive contamination has a profound and long-term effect on the environment. Radioactive Cs 137 represents a serious environmental problem in Japan following the release of large amounts of radioactive material from the Fukushima Daiichi nuclear power plant during an incident caused indirectly by the Great East Japan Earthquake in 2011. As the half-life of Cs 137 is 30.1 years, it can be considered a long-lasting environmental pollutant in local ground soils and seawater. Currently, the only effective means for the removal of radiocesium from a contaminated area is large-scale excavation of surface soil, which has been accomplished over large swathes of land in the vicinity of the Fukushima Daiichi Nuclear Power Plant. Decontamination projects from soil are difficult because soil clays have a strong adsorptive capacity. Safe and effective sensing of radioactive materials that are present as contaminating agents is required. The characteristic gamma rays emitted by radioactive isotopes enable the detection of several isotopes, although it is difficult to visualize these emissions over small regions and under highly radioactive background conditions. Therefore, it would be useful if the location of Cs (or radiocesium) could be visualized to trace leaks or other processes from facilities containing high-level radioactive materials, and to reduce the volume of radioactively contaminated materials generated during the decommissioning of a nuclear reactor. With this in mind, supramolecular approaches for environmental sensing may lead to resolutions higher than the micrometer level. A chemical optical probe provides higher spatial resolution and is less expensive than existing radioscopes and gamma-ray cameras.
We have developed a fluorescent molecular probe with excellent selectivity for cesium ions, called “Cesium Green” bearing an ethylene glycol moiety for selective capture of Cs ions and an oligo phenol moiety for fluorescence output 53) (Fig. 10). Cesium Green emitted green fluorescence in the presence of Cs ions, whereas blue emission was observed with lithium, sodium, potassium, and rubidium ions (Fig. 10). Cesium Green has a higher spatial resolution for cesium ions than existing radioscopes and gamma ray cameras because only contaminated particles are detected by the eye and can be easily removed, and the volume of radioactively contaminated material for extraction is greatly reduced (Fig. 10). Because the location of cesium in solid samples such as soil, foodstuffs, and plants can be visualized using this probe, we can study its diffusion processes and accumulation behavior in these media. The availability of this and other similar easily implemented tests for environmental contaminants is likely to increase the volume of data regarding rates of contamination around chemical and radiological hazards. This information will be useful for constructing maps of contamination that can be used to determine accommodation and agricultural policies involving local populations.

Visualization of Cs ions on the solid surface, in the cell, and in an aqueous medium by using the newly developed optical probes.
We believe that micrometer-scale imaging of Cs particles could be extended to cellular-level imaging. Cs uptake by plants occurs via the potassium transport system, and the resulting intracellular Cs distribution in plants is of interest, although an imaging method has not yet been established. Determination of the intracellular localization of Cs in plants is particularly important for phytoremediation or for the detection of plants contaminated with Cs. Therefore, we applied cesium green to plant cell imaging using Arabidopsis cotyledons. In this study, Cs imaging was performed using plants treated with high concentrations of Cs. Arabidopsis seeds were germinated and grown for 9 days on a medium containing 0.5 mM cesium carbonate. Elemental analyses revealed a Cs+ content in the resulting plants of 67±11 nmol Cs/mg dry weight. The resulting seedlings were freeze-dried and soaked in a 0.02 wt% methanolic Cesium Green solution prior to imaging by fluorescence microscopy.
Upon comparison between the fluorescence images of Cs-treated and non-treated plants, bright fluorescent spots were visible in the cotyledons of the Cs-treated plants, but not in the untreated plants 54) (Fig. 10). It is likely that these fluorescent spots are vacuoles, which play a role in the detoxification of potentially toxic substances (such as sodium under salt stress) by sequestration. It is possible that plants sequester Cs ion in vacuoles to avoid negative effects in the cytoplasm. From this result, it is apparent that cesium green is suitable for the detection of Cs in plants at the organelle level. This may contribute to a better understanding of the underlying mechanisms.
The newly developed fluorescence probe Cesium Green selectively emitted green fluorescence in the presence of Cs ions and successfully demonstrated the visualization of Cs ions in freeze-dried cells of Arabidopsis. However, despite its excellent selectivity, Cesium Green shows no recognition of Cs ions in water or other polar media because of its poor coupling ability. We developed an optode membrane with high selectivity toward Cs ions under typical environmental conditions in domestic water supply or seawater 55) . The optode membrane is especially suitable for detecting Cs ions and promoting their ability to detect them with the naked eye in environmental water. Nano-optode particles with a diameter of ca. 100 nm were successfully prepared by sonication of the optode membrane. The nano-optode showed an excellent response to Cs ions at sub-micromolar levels (Fig. 10). The optode is also potentially applicable to biological systems, so that the diffusion processes and accumulation behavior of Cs ions in the environment can be studied.
The inner and interfacial environments of self-assemblies formed by amphiphiles provide fascinating nano-spaces for selective and efficient chemical reactions and processes. Based on these advantages, we demonstrated (a) photo-induced rapid controlled release using the inner environments of molecular assemblies and (b) recognition and sensing of anions and cations using the inside of self-assembled molecular films at the interfaces. Advances in the development of molecular functional systems are expected based not only on molecular structures but also on the spaces and environments produced by them.
The soft and flexible supramolecular nano-spaces show dynamic changes in their properties by external stimuli, such as applications of light irradiation and electric field, and inclusions of ions and molecules. To enhance their functions, now we work on the analysis of the dynamics of their functional changes by using in-situ measurement systems and feedback on the design of structures of the amphiphiles and molecular assemblies. We are trying to understand the dynamics of the hierarchical processes. Furthermore, to avoid damage to the drugs, embedded in the photoresponsive molecular assemblies, and organisms by ultraviolet light irradiation, we are developing molecular assemblies responding to red or near-infrared light that penetrates the biological tissues. Moreover, we investigate the conversion of molecular recognition event at the interfaces into changes in the functions, e.g. wettability and signal transduction through the electric properties unlike conventional applications, such as sensors and imaging. Our research dream is the development of a new molecular recognition system including the controlled release by using the inner and interfacial environments of self-assemblies.
I am grateful to thank prof. Hideki Sakai and prof. Kenichi Sakai (Tokyo University of Science) for their supervision and support. I would like to thank past and current members of Sakai and Sakai group of Tokyo University of Science: Mr. Kazuki Kobayashi, Mr. Koji Yamanaga, Ms. Ayumi Kimura, Ms. Yurina Kanehara, Mr. Kohei Tanaka, Mr. Tatsuki Morita, and Ms. Risa Tanaka for their enthusiastic effort in these projects. I am grateful to thank Dr. Hiroki Iwase (CROSS) for the collaborative work in neutron scattering measurements. This research was partly supported by the Foundation, Oil & Fat Industry Kaikan and JSPS KAKENHI Grant-in-Aid for Early-Career Scientists (Grant Number JP20K15248).