Abstract
Juvenile hormone (JH) agonists constitute a subclass of insect growth regulators and play important roles in insect pest management. In this work, a multi-step virtual screening program was executed to find novel JH agonists. A database of 5 million purchasable compounds was sequentially processed with three computational filters: (i) shape and chemical similarity as compared to known JH-active compounds; (ii) molecular docking simulations against a Drosophila JH receptor, methoprene-tolerant; and (iii) free energy calculation of ligand–receptor binding using a modified MM/PBSA (molecular mechanics/Poisson–Boltzmann surface area) protocol. The 11 candidates that passed the three filters were evaluated in a luciferase reporter assay, leading to the identification of a hit compound that contains a piperazine ring system (EC50=870 nM). This compound is structurally dissimilar to known JH agonists and synthetically easy to access; therefore, it is a promising starting point for further structure optimization.

Introduction
Juvenile hormones (JHs) are acyclic sesquiterpenoids that regulate various aspects of insect physiology.1) A principal role of this hormone is preventing metamorphosis induced by the molting hormone (20-hydroxyecdysone), which allows insects to proceed to larval molting. Because JHs are not present in animals other than arthropods, synthetic JH agonists (also referred to as juvenoids) are promising candidates for insecticides with reduced safety concerns. Shortly after the structure determination of JH I2) (Fig. 1), synthetic efforts by Zoecon chemists led to the discovery of the first-in-class juvenoid insecticide methoprene, which was followed by fenoxycarb (Dr. R. Maag/Roche) and pyriproxyfen (Sumitomo Chemical).3) These juvenoids were discovered through analog synthesis or random screening; no JH agonist has hitherto been developed via a rational, computational approach.
After the discovery of juvenoids, substantial effort has been made to elucidate their molecular mode of action. Through a mutagenesis screen in Drosophila melanogaster, Wilson et al.4,5) identified a bHLH/PAS (basic helix-loop-helix/Per-Arnt-Sim) protein termed methoprene-tolerant (Met), whose loss confers resistance to lethal doses of methoprene. Miura et al.6) found that the D. melanogaster Met (DmMet) is capable of binding JHs with nanomolar affinities; this finding was later extended to Met proteins of other insects.7,8) Furthermore, the liganded Met proteins were shown to heterodimerize with another bHLH/PAS protein, named taiman (Tai),9) and activate the transcription of early response genes, such as early trypsin10) and Krüppel homolog 1.11) These pioneering studies have established the Met/Tai complex as the intracellular receptor of JH agonists.12,13) Although the three-dimensional structure of Met remains to be solved, X-ray crystal structures of related bHLH/PAS proteins can be used to construct structural models of the JH receptor. In fact, Charles et al.7) combined homology modeling and site-directed mutagenesis to identify amino acid residues of Tribolium castaneum Met (TcMet) that are important for JH binding.
In this work, we conducted a hierarchical virtual screening program to identify novel JH agonists. This program consists of three steps: (i) ligand-based virtual screening using known JH agonists (1 and 2, Fig. 1) as the template; (ii) docking screening against the DmMet homology model; and (iii) molecular dynamics (MD)-based MM/PBSA14) (molecular mechanics/Poisson–Boltzmann surface area) rescoring corrected by the ligand conformational free energy calculation.15) From a database of 5 million compounds, 11 compounds were selected and subjected to a luciferase reporter assay in HEK293T cells that were transfected with Drosophila Met and Tai genes.16) We successfully identified a novel piperazine-based JH agonist with submicromolar potency.
Materials and methods
1. Virtual screening1.1. DatabaseA structure database of 5 million commercially available compounds was provided by Namiki Shoji (Tokyo, Japan). This database was processed with OMEGA 2.5.1.417,18) to generate a maximum of 200 conformers per compound.
1.2. Shape-based screeningShape-based screening was performed using ROCS 3.2.2.19,20) The query was generated from two known JH agonists, 2-[2-(4-phenoxyphenoxy)ethoxy]thiazole (1) and 2-[3-(4-phenoxyphenoxy)-1-propyn-1-yl)pyridine (2), using the Implicit Mills–Dean color force field. The TanimotoCombo score was calculated for each member of the database, and the top 1,000 compounds were selected for the docking screening.
1.3. Homology modelingThe three-dimensional structure of the DmMet PAS-B domain was constructed on Isolated-FAMSD system.21,22) The primary sequence of the target protein was extracted from the full-length DmMet sequence4) (GenBank accession code: AAC14350). The X-ray crystal structure of the Mus musculus Period circadian protein homolog 1 (Per1; PDB code: 4DJ2) was used as the template for the homology modeling. A Ramachandran plot of the homology model is shown in Fig. S1. The internal cavity was detected using the CASTp 3.0 web server,23) and structural figures were generated using UCSF Chimera 1.14.24)
1.4. Docking screeningDocking screening was performed using OEDocking 3.2.0.2.25) The MakeReceptor module was used to define the ligand-binding site of the Drosophila Met homology model. The FRED module26,27) was used to dock the 1,000 candidates into the binding site, and the docking poses were ranked using the Chemgauss4 scoring function. The top 120 compounds were subjected to visual inspection based on the novelty of their chemical scaffolds, and 50 compounds were selected for MD-based MM/PBSA screening.
1.5. MD-based MM/PBSA screeningMD-based MM/PBSA screening was conducted using Amber1628) and SZYBKI 1.8.0.129) according to a modified method described in the literature.15) The ligand–receptor complexes obtained from the docking screening were used as the initial structures. The atomic partial charges of the ligands were assigned using the AM1-BCC method30,31) implemented in the antechamber module of Amber16. The simulation systems were prepared using the tleap module of Amber16. The GAFF2 and ff14SB32) force fields were used to describe the ligands and the receptor, respectively. The net charge of the systems was neutralized by adding Na+ and Cl− ions, and the systems were solvated in a TIP3P water box33) that extends at least 10 Å from the solute surface.
MD simulations were performed using the pmemd.cuda module of Amber16. Periodic boundary conditions were used for all simulations. Electrostatic interactions were calculated using the particle mesh Ewald method34) with a cutoff of 12.0 Å. Covalent bonds to hydrogens were restrained by the SHAKE algorithm.35) The time step was set to 1 fs. Before the MD simulations, the systems were energy-minimized using the steepest descent and conjugate gradient algorithms. The systems were then gradually heated from 0 to 300 K over 50 ps in a canonical (NVT) ensemble and equilibrated for 900 ps in an isothermal–isobaric (NPT) ensemble. Production simulations were performed for 5 ns in an NPT ensemble, during which the snapshots were sampled every 25 ps; thus, 200 snapshots were collected in each simulation.
MM/PBSA calculations were performed using the MMPBSA.py.MPI module of Amber16. The internal and external dielectric constants were set to 1.0 and 80.0, respectively. The ionic strength was set to 0.2 mM. The MM/PBSA scores were corrected by the ligand conformational free energy terms, which were calculated using the FREEFORM module36) of SZYBKI. Although the MM/PBSA calculation was performed against 200 snapshots/compound, the ligand conformational free energy calculation was applied to 20 snapshots/compound to reduce the computational cost.
2. Reporter gene assayThe reporter gene assay was performed as described in our earlier report.16) HEK293T cells were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin at 37°C under a 5% CO2 atmosphere. The cell suspension (400 µL/well) was seeded in a 24-well plate at a density of 0.5–1.0×105 cells/mL and incubated for 2 days. After removal of the growth medium, each well received 200 µL of the transfection mixture that contained pCS2+DmMet (100 ng), pCS2+DmTai (200 ng), pGL3-BmkJHRE×2-Fluc (180 ng), pRL-SV40 (18 ng), and the TransFast Transfection Reagent (1.5 µL; Promega, Madison, WI, USA). The cells were incubated for 1 hr, diluted with 400 µL of DMEM, and incubated for an additional 24 hr. The cells were then treated with 6 µL of a test compound solution in dimethyl sulfoxide. After incubation for 24 hr, the reporter activity was measured using the Dual-Luciferase Reporter Assay System (Promega) in accordance with the manufacturer’s protocol. The firefly luciferase activity was normalized against the Renilla luciferase activity to yield the relative luminescence unit (RLU). The half-maximal effective concentrations (EC50, M) were determined via probit regression analyses using PriProbit 1.63.37)
3. Chemical characterization of compound 13Compound 13 was from Enamine (Kyiv, Ukraine), and its purity is guaranteed to be >90%. The structure was confirmed via 1H NMR and high-resolution mass spectrometry (HRMS) using Bruker Avance III 400 (Billerica, MA, USA) and Thermo Fisher Scientific Exactive Plus (Waltham, MA, USA) spectrometers, respectively. 1H NMR (400 MHz, 298 K, CDCl3; Fig. S2): δ 7.96–7.90 (m, 2H), 6.86–6.79 (m, 2H), 5.05–3.98 (br m, 2H), 3.87 (s, 3H), 3.82–3.10 (br m, 4H), 3.03 (br m, 1H), 1.75 (m, 1H), 1.50–1.20 (br m, 3H), 1.09–0.97 (m, 2H), 0.86–0.75 (m, 2H). HRMS–ESI (m/z): calcd for C17H23N2O3 [M+H]+, 303.1703; found, 303.1705.
Results and discussion
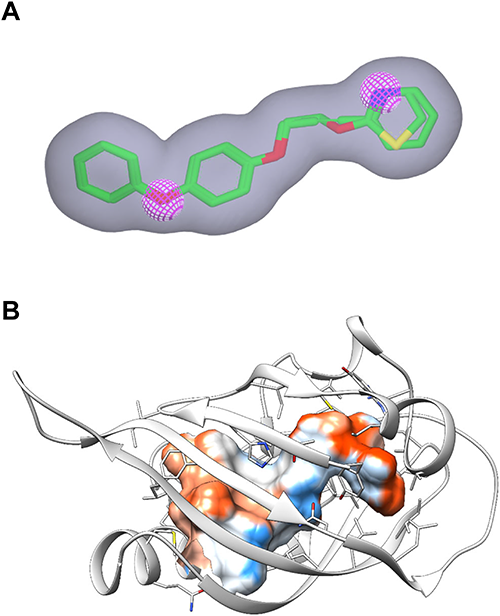
To identify novel JH agonists, we performed a multi-step virtual screening cascade against a database of 5 million purchasable compounds (Fig. 2). The first screening was performed using the OpenEye tool ROCS, which calculates shape and chemical (e.g., hydrogen-bond donor/acceptor, ring, hydrophobe, etc.) similarity of compounds to a query generated from known active compounds. We used compounds 1 and 2 for the query generation because these juvenoids outperformed natural JHs in our reporter system.16) The initial query generated automatically by ROCS contained six chemical features shared by these two juvenoids: i.e., three ring features (corresponding to two benzene rings and one thiazole/pyridine ring) and three hydrogen-bond acceptor features (corresponding to two ether oxygens and one thiazole/pyridine nitrogen). We removed all of the ring features and one hydrogen-bond acceptor feature to increase the structural diversity of hit compounds (Fig. 3A). All compounds in the database were ranked according to their TanimotoCombo scores, and the top 1,000 compounds were selected for the second screening.
The second screening was done using the FRED program as the docking engine. We selected DmMet as the target for this screening because our reporter system was built on the DmMet/DmTai complex.16) Before the docking screening, we first constructed a homology model of the DmMet PAS-B domain using the X-ray structure of the mouse Per1 protein as the template. Although the sequence identity between the target and template proteins was only 20%, this was the best-matched pair at the time of modeling. The generated model contained an internal cavity surrounded mainly by hydrophobic amino acid residues (Fig. 3B), which is suitable for binding lipophilic ligands such as JHs. The 1,000 compounds that passed the first screening were docked into the cavity and ranked according to the Chemgauss4 scoring function. The top-scoring 120 compounds, whose docking scores ranged from −17 to −13, were visually inspected for the novelty of their chemical scaffolds. Thus, 50 compounds were selected for a third screening. Among these, we purchased 7 compounds (3–9) for assessing the reliability of the Chemgauss4 scoring.
The third screening was conducted using our MM/PBSA protocol,15) where the MM/PBSA calculation of the MD trajectory is coupled to the ligand conformer free energy estimation with FREEFORM. We performed a 5 ns MD simulation for each ligand–receptor complex selected in the second screening and extracted 200 snapshots for each complex. We then subjected these snapshots to our modified MM/PBSA calculations and estimated the free energy of ligand–receptor binding (ΔGbind). Compounds 3–9, which were selected based on their docking scores, showed a wide range of ΔGbind values (Table 1), and some of them were predicted to be non-binders (i.e., ΔGbind>0). We purchased four additional compounds (10–13) based on their ΔGbind scores. Although stereoisomers were treated as separate compounds in the virtual screening, the purchased compounds, except for 4, were mixtures of stereoisomers.
We submitted the 11 purchased candidates to the reporter assay in HEK293T cells16) (Table 1). The compounds were first assessed in single-dose testing at 100 µM, and only compound 13 increased the reporter activity. As shown in Fig. 4A, this compound induced clear dose-dependent activation of the luciferase gene. The EC50 value of 13 was 0.87 µM, which is 140 times less potent than JH I (Table 1). However, compound 13 (CLogP=1.28) is several orders of magnitude less hydrophobic than commercialized juvenoids (methoprene, log P=5.00; fenoxycarb, log P=4.07; pyriproxyfen, log P=5.37).38) In conjunction with the lipophilic nature of the binding site (Fig. 3B), this compound leaves much room for improving potency. In addition, the simple chemical scaffold of 13 makes optimization of its structure easy.
Figure 4B shows a representative docking pose of compound 13 extracted from the MD trajectory. Note that the (S)-isomer of 13 is docked into the binding site, which means that the (R)-isomer was filtered out during the virtual screening cascade. In this model, compound 13 is accommodated in the hydrophobic cavity and is hydrogen-bonded to two Tyr residues, Y404 and Y468, via its ester carbonyl and amide carbonyl oxygens, respectively. As shown in Fig. S3, these hydrogen bonds are relatively stable during the simulation, although some fluctuations were visible at the beginning. Thus, Y404 and Y468 of DmMet are likely to play crucial roles in recognizing compound 13. A sequence alignment revealed that these Tyr residues are well conserved across insects of different taxonomic orders, including Diptera, Lepidoptera, Coleoptera, Hemiptera, and Orthoptera (Fig. S4). Mutagenesis experiments against the DmMet paralog, germ cell-expressed (DmGce), revealed that Y270 in DmGce, which corresponds to Y404 in DmMet, is important for JH recognition, and docking simulations predicted a hydrogen bond between the hydroxy group of Y270 and the epoxide of JHs.39) Thus, the ester group of 13 is likely to act as a surrogate for the epoxide moiety of JHs. To test this hypothesis and improve potency, a structure–activity relationship study of 13 is currently in progress.
In this study, the 7 compounds selected based on the Chemgauss4 scores (3–9) were not active in the reporter assay. By contrast, one of the four compounds selected in the modified MM/PBSA rescoring (10–13) significantly induced luciferase activity. Thus, our MM/PBSA protocol worked well for screening JH agonists. This is the third example where our MM/PBSA approach showed its effectiveness: the first was the virtual screening of nonsteroidal ecdysone agonists,15) and the second was the rational design of NSBR1, a nonsteroidal brassinolide-like compound.40) A major limitation of this MM/PBSA protocol is its relatively high computational cost; it cannot be used to screen millions of compounds. Nevertheless, as demonstrated in this study, incorporating this protocol into late-stage virtual screening improves the rate of screening success. We are now attempting to further improve the prediction accuracy of this MM/PBSA approach and expand its applicability, which may be reported in due course.
Conclusion
To discover novel JH agonists, we have carried out a hierarchical virtual screening campaign that consists of (i) shape-based screening using ROCS, (ii) docking screening using FRED, and (iii) modified MM/PBSA rescoring using Amber and FREEFORM. Among the 11 candidates selected, piperazine derivative 13 was successfully identified as a novel JH agonist. The submicromolar potency, moderate hydrophobicity, and simple scaffold of 13 make this compound a promising starting point for further structure optimization.
Acknowledgments
We are grateful to Dr. K. Komatsu (In-Silico Sciences) and Dr. H. Umeyama (Professor Emeritus at Kitasato University) for their help in protein modeling. We also thank OpenEye Scientific Software for providing us with an academic license. Finally, we would like to thank K. Nishimura, M. Ueno, and B. Nishikawa (Kyoto University) for supporting our spectrometric analyses. This work was supported in part by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Numbers JP16K07625 (to YN), JP17J01486 (to TY), and JP19K06051 (to YN). TY was supported by the JSPS Research Fellowship for Young Scientists.
Electronic supplementary materials
The online version of this article contains supplementary materials (Supplemental Figs. S1–S4), which are available at http://www.jstage.jst.go.jp/browse/jpestics/.
References
- 1) W. G. Goodman and M. Cusson: The Juvenile Hormones. In “Insect Endocrinology,” ed. by L. I. Gilbert, Elsevier, Amsterdam, pp. 310–365 (2012).
- 2) H. Röller, K. H. Dahm, C. C. Sweely and B. M. Trost: The structure of the juvenile hormone. Angew. Chem. Int. Ed. Engl. 6, 179–180 (1967).
- 3) T. S. Dhadialla, G. R. Carlson and D. P. Le: New insecticides with ecdysteroidal and juvenile hormone activity. Annu. Rev. Entomol. 43, 545–569 (1998).
- 4) M. Ashok, C. Turner and T. G. Wilson: Insect juvenile hormone resistance gene homology with the bHLH-PAS family of transcriptional regulators. Proc. Natl. Acad. Sci. U.S.A. 95, 2761–2766 (1998).
- 5) T. G. Wilson and M. Ashok: Insecticide resistance resulting from an absence of target-site gene product. Proc. Natl. Acad. Sci. U.S.A. 95, 14040–14044 (1998).
- 6) K. Miura, M. Oda, S. Makita and Y. Chinzei: Characterization of the Drosophila Methoprene-tolerant gene product. FEBS J. 272, 1169–1178 (2005).
- 7) J.-P. Charles, T. Iwema, V. C. Epa, K. Takaki, J. Rynes and M. Jindra: Ligand-binding properties of a juvenile hormone receptor, Methoprene-tolerant. Proc. Natl. Acad. Sci. U.S.A. 108, 21128–21133 (2011).
- 8) M. Li, P. Liu, J. D. Wiley, R. Ojani, D. R. Bevan, J. Li and J. Zhu: A steroid receptor coactivator acts as the DNA-binding partner of the methoprene-tolerant protein in regulating juvenile hormone response genes. Mol. Cell. Endocrinol. 394, 47–58 (2014).
- 9) J. Bai, Y. Uehara and D. J. Montell: Regulation of invasive cell behavior by Taiman, a Drosophila protein related to AIB1, a steroid receptor coactivator amplified in breast cancer. Cell 103, 1047–1058 (2000).
- 10) M. Li, E. A. Mead and J. Zhu: Heterodimer of two bHLH-PAS proteins mediates juvenile hormone-induced gene expression. Proc. Natl. Acad. Sci. U.S.A. 108, 638–643 (2011).
- 11) T. Kayukawa, C. Minakuchi, T. Namiki, T. Togawa, M. Yoshiyama, M. Kamimura, K. Mita, S. Imanishi, M. Kiuchi, Y. Ishikawa and T. Shinoda: Transcriptional regulation of juvenile hormone-mediated induction of Krüppel homolog 1, a repressor of insect metamorphosis. Proc. Natl. Acad. Sci. U.S.A. 109, 11729–11734 (2012).
- 12) M. Jindra, X. Bellés and T. Shinoda: Molecular basis of juvenile hormone signaling. Curr. Opin. Insect Sci. 11, 39–46 (2015).
- 13) M. Jindra and L. Bittova: The juvenile hormone receptor as a target of juvenoid “insect growth regulators”. Arch. Insect Biochem. Physiol. 103, e21615 (2020).
- 14) P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case and T. E. Cheatham 3rd: Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 33, 889–897 (2000).
- 15) S. Horoiwa, T. Yokoi, S. Masumoto, S. Minami, C. Ishizuka, H. Kishikawa, S. Ozaki, S. Kitsuda, Y. Nakagawa and H. Miyagawa: Structure-based virtual screening for insect ecdysone receptor ligands using MM/PBSA. Bioorg. Med. Chem. 27, 1065–1075 (2019).
- 16) T. Yokoi, T. Nabe, C. Ishizuka, K. Hayashi, S. Ito-Harashima, T. Yagi, Y. Nakagawa and H. Miyagawa: Transcription-inducing activity of natural and synthetic juvenile hormone agonists through the Drosophila Methoprene-tolerant protein. Pest Manag. Sci. 76, 2316–2323 (2020).
- 17) OMEGA 2.5.1.4, OpenEye Scientific Software, Inc., Santa Fe, NM, USA.
- 18) P. C. D. Hawkins, A. G. Skillman, G. L. Warren, B. A. Ellingson and M. T. Stahl: Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 50, 572–584 (2010).
- 19) ROCS 3.2.2, OpenEye Scientific Software, Inc., Santa Fe, NM, USA.
- 20) P. C. D. Hawkins, A. G. Skillman and A. Nicholls: Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 50, 74–82 (2007).
- 21) Isolated-FAMSD, In-Silico Sciences, Inc., Tokyo, Japan.
- 22) K. Ogata and H. Umeyama: An automatic homology modeling method consisting of database searches and simulated annealing. J. Mol. Graph. Model. 18, 258–272, 305–306 (2000).
- 23) W. Tian, C. Chen, X. Lei, J. Zhao and J. Liang: CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 46(W1), W363–W367 (2018).
- 24) E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin: UCSF Chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
- 25) OEDocking 3.2.0.2, OpenEye Scientific Software, Inc., Santa Fe, NM, USA.
- 26) M. McGann: FRED Pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 51, 578–596 (2011).
- 27) M. McGann: FRED and HYBRID docking performance on standardized datasets. J. Comput. Aided Mol. Des. 26, 897–906 (2012).
- 28) D. A. Case, R. M. Betz, D. S. Cerutti, T. E. Cheatham III, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, C. Lin, T. Luchko, R. Luo, B. Madej, D. Mermelstein, K. M. Merz, G. Monard, H. Nguyen, H. T. Nguyen, I. Omelyan, A. Onufriev, D. R. Roe, A. Roitberg, C. Sagui, C. L. Simmerling, W. M. Botello-Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, L. Xiao and P. A. Kollman: AMBER 2016, University of California, San Francisco, CA, USA.
- 29) SZYBKI 1.8.0.1, OpenEye Scientific Software, Inc., Santa Fe, NM, USA.
- 30) A. Jakalian, B. L. Bush, D. B. Jack and C. I. Bayly: Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 21, 132–146 (2000).
- 31) A. Jakalian, D. B. Jack and C. I. Bayly: Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 23, 1623–1641 (2002).
- 32) J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling: ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015).
- 33) W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein: Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
- 34) U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen: A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995).
- 35) J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen: Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 (1977).
- 36) S. Wlodek, A. G. Skillman and A. Nicholls: Ligand entropy in gas-phase, upon solvation and protein complexation. Fast estimation with quasi-Newton hessian. J. Chem. Theory Comput. 6, 2140–2152 (2010).
- 37) M. Sakuma: Probit analysis of preference data. Appl. Entomol. Zool. 33, 339–347 (1998).
- 38) https://sitem.herts.ac.uk/aeru/ppdb/en/index.htm (Accessed 27 Sep., 2020).
- 39) L. Bittova, P. Jedlicka, M. Dracinsky, P. Kirubakaran, J. Vondrasek, R. Hanus and M. Jindra: Exquisite ligand stereoselectivity of a Drosophila juvenile hormone receptor contrasts with its broad agonist repertoire. J. Biol. Chem. 294, 410–423 (2019).
- 40) A. Sugiura, S. Horoiwa, T. Aoki, S. Takimoto, A. Yamagami, T. Nakano, Y. Nakagawa and H. Miyagawa: Discovery of a nonsteroidal brassinolide-like compound, NSBR1. J. Pestic. Sci. 42, 105–111 (2017).
- 41) J. Kyte and R. F. Doolittle: A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132 (1982).