Abstract
Herein, we provide a brief overview of the synthesis and applications of trifluoromethylpyridine (TFMP) and its derivatives in the agrochemical and pharmaceutical industries. Currently, the major use of TFMP derivatives is in the protection of crops from pests. Fluazifop-butyl was the first TFMP derivative introduced to the agrochemical market, and since then, more than 20 new TFMP-containing agrochemicals have acquired ISO common names. Several TFMP derivatives are also used in the pharmaceutical and veterinary industries; five pharmaceutical and two veterinary products containing the TFMP moiety have been granted market approval, and many candidates are currently undergoing clinical trials. The biological activities of TFMP derivatives are thought to be due to the combination of the unique physicochemical properties of the fluorine atom and the unique characteristics of the pyridine moiety. It is expected that many novel applications of TFMP will be discovered in the future.
Introduction
Many recent advances in the agrochemical, pharmaceutical, and functional materials fields have been made possible by the development of organic compounds containing fluorine. Indeed, the effects of fluorine and fluorine-containing moieties on the biological activities and physical properties of compounds have earned fluorine a unique place in the arsenal of the discovery chemist. As the number of applications for these compounds continues to grow, the development of fluorinated organic chemicals is becoming an increasingly important research topic.
In the crop protection industry, more than 50% of the pesticides launched in the last two decades have been fluorinated. In addition, around 40% of all fluorine-containing pesticides currently on the market contain a trifluoromethyl group, making these compounds an important subgroup of fluorinated compounds.1) The biological activities of fluorine-containing compounds are considered to be derived from the unique physicochemical properties of fluorine (van der Waals radius, 1.47 Å),2) which, sterically, is the next smallest atom after hydrogen (van der Waals radius, 1.20 Å)2) but the atom with the largest electronegativity (3.98).3) In addition, because the carbon–fluorine bond is relatively short (1.38 Å) compared with the other carbon–halogen bonds, the bond has strong resonance. As a result, the Hammett constant (σp) of fluorine is 0.06,4) which is similar to that of hydrogen. Interestingly, the electronegativity of the trifluoromethyl group is 3.46,5) and its Hammett constant is 0.54,4) indicating that, unlike fluorine, the trifluoromethyl group is strongly electron withdrawing. Therefore, during compound development, the trifluoromethyl group can be treated as a purely electron-withdrawing group.
These unique properties of fluorine mean that substitution with a fluorine or fluorine-containing moiety can have a large impact on the conformation, acid dissociation constant, metabolism, translocation, and biomolecular affinity of a compound. This has meant that bioisosteric replacement of hydrogen with fluorine has become a useful means of designing compounds with unique biological properties. For similar reasons, much effort has been made to develop synthetic methods for introducing trifluoromethyl groups into aromatic rings. The first synthesis of an aromatic compound bearing a trifluoromethyl group was reported in 1898 by Swarts,6) who treated benzotrichloride with antimony trifluoride to afford benzotrifluoride; the same transformation using hydrogen fluoride was subsequently achieved under liquid-phase reaction conditions in the 1930s.7) In 1947, the introduction of a trifluoromethyl group into a pyridine ring to afford trifluoromethylpyridine (TFMP) using a synthetic procedure similar to that used for benzotrifluoride but involving chlorination and fluorination of picoline (Scheme 1) was first reported.8) Comparing the physicochemical properties of TFMP and benzotrifluoride, there is a significant difference in the hydrophobic constant (e.g., 3-(trifluoromethyl)pyridine 1.7 versus benzotrifluoride 3.0), which can be expected to provide TFMP-containing compounds with many advantages, such as novel biological activity, lower toxicity, and advanced systemic and/or good selectivity; therefore, many efforts have been made to achieve the synthesis of TFMP. However, to make enough TFMP for use as a raw material for industrial production, it is important to establish a practical large-scale industrial manufacturing process. Details of the industrial manufacturing of TFMP and its use in the manufacture of various agrochemicals and pharmaceuticals are discussed in this review.
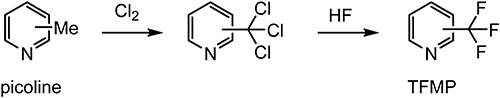
Scheme 1. Liquid-phase synthesis of TFMP.
1. Demand for TFMP isomers and development of TFMP derivatives
1.1. Trends in the demand for TFMP isomers
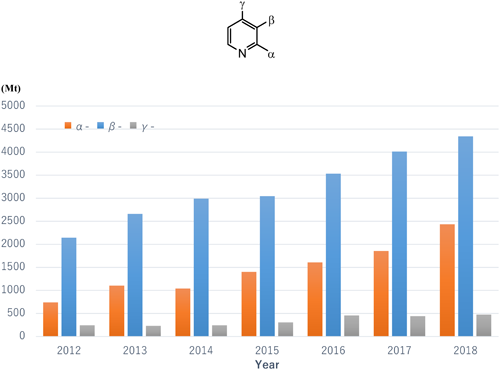
Figure 1 shows the worldwide demand for TFMP isomers used as intermediates in the production of synthetic pesticides for the period from 2012 to 2018.
The production volume of each TFMP isomer (Fig. 1) was estimated based on the following data sources: the sales volume of each formulated agrochemical, which was obtained from i-map Sigma (https://kynetecwebsc.com/documentation/i-map/3.29.0/), a database for the crop protection market provided by the market research company Kynetec (Newbury, UK); the concentration of each active ingredient in the formulated product; and the synthetic yield described in the patent for each agrochemical containing a TFMP moiety.
Every year from 2012 to 2018, the demand was greatest for β-TFMP, followed by α-TFMP and γ-TFMP. In addition, the demand for each of the three TFMP isomers increased each year. Examining the sales of the pesticides individually, globally in 2018, fluazinam and haloxyfop were the two top-selling pesticides possessing the β-TFMP moiety. In addition, the total sales volumes of fluopicolide and fluopyram, which also contain the β-TFMP moiety, have gradually increased from 2014 to 2018 and are now around 1,000 tons/year. Picoxystrobin is the only pesticide manufactured using the α-TFMP intermediate with sales of more than 2,000 tons/year. However, sales of bicyclopyrone have markedly increased in the last few years, which has increased demand for α-TFMP. Around 500 tons/year of pesticides containing the γ-TFMP intermediate are manufactured; therefore, the demand for γ-TFMP is relatively small.
1.2. Research and development of TFMP derivatives
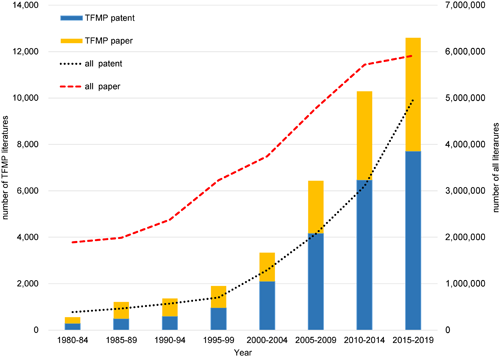
Research and development activities (i.e., the outputs of scientific papers and patents) involving TFMP derivatives from 1980 to 2019 were examined using data obtained through crossover analysis of the STN International Registry9) and HCAplus databases10) (CAS, Columbus, Ohio, USA, and FIZ Karlsruhe, Eggenstein-Leopoldshafen, Germany). Since the development of economically feasible processes for the synthesis of several TFMP intermediates from 3-picoline in the early 1980s, research and development activity involving TFMP derivatives has rapidly and consistently increased each year (Fig. 2).
2. Synthesis of TFMP derivatives
There are three main methods for preparing TFMP derivatives: chlorine/fluorine exchange using trichloromethylpyridine; construction of a pyridine ring from a trifluoromethyl-containing building block; or direct introduction of a trifluoromethyl group using a trifluoromethyl active species such as trifluoromethyl copper, which undergoes substitution reactions with bromo- and iodopyridines.11) The first two methods are currently the most commonly used; therefore, the discussion below focuses on those two methods.
2.1. Chlorine/fluorine exchange using trichloromethylpyridine
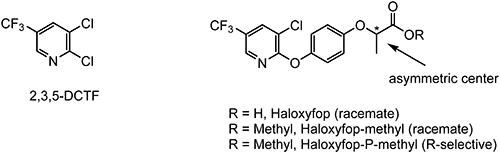
Among TFMP derivatives, 2,3-dichloro-5-(trifluoromethyl)pyridine (2,3,5-DCTF), which is used as a chemical intermediate for the synthesis of several crop-protection products, is in the highest demand (production data estimated from the i-map Sigma database). Various methods of synthesizing 2,3,5-DCTF have been reported. For example, 2-chloro-5-methylpyridine or 2-chloro-5-(chloromethyl)pyridine can be chlorinated under liquid-phase conditions to afford the intermediate 2,3-dichloro-5-(trichloromethyl)pyridine (2,3,5-DCTC); subsequent vapor–phase fluorination of 2,3,5-DCTC produces 2,3,5-DCTF (Scheme 2).12–14)
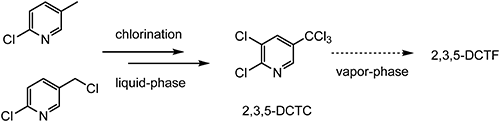
Scheme 2. Stepwise liquid-phase/vapor–phase synthesis of 2,3,5-DCTF.
An approach using stepwise vapor–phase chlorination followed by fluorination has also been reported (Scheme 3).15,16)
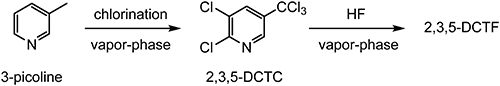
Scheme 3. Stepwise vapor–phase synthesis of 2,3,5-DCTF.
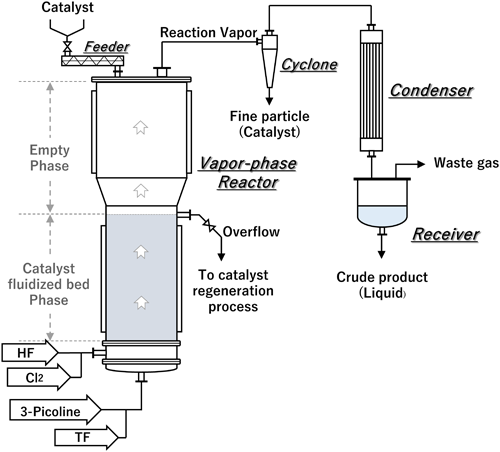
Another well-known approach is simultaneous vapor–phase chlorination/fluorination at a high temperature (>300°C) with transition metal-based catalysts such as iron fluoride (Scheme 4).17,18) The simultaneous vapor–phase reaction has the advantage that 2-chloro-5-(trifluoromethyl)pyridine (2,5-CTF), a key intermediate for the synthesis of fluazifop, can be obtained in good yield via a simple one-step reaction. The number of chlorine atoms introduced to the pyridine ring can be controlled by changing the molar ratio of chlorine gas and the reaction temperature; however, the formation of some multi-chlorinated by-products is unavoidable. Fortunately, these unwanted by-products can be reduced to 3-(trifluoromethyl)pyridine (3-TF) by catalytic hydrogenolysis and then fed back into the reactor to reduce overall production costs.
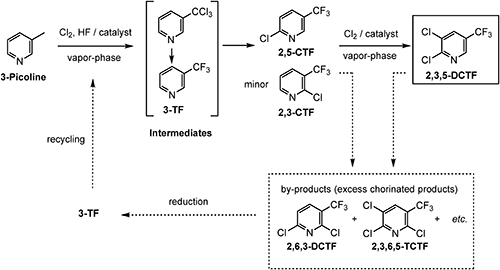
Scheme 4. Simultaneous vapor–phase synthesis of TFMPs.
The vapor–phase reactor used for this approach includes two phases: a catalyst fluidized-bed phase and an empty phase (Fig. 3). In the fluidized-bed phase, fluorination proceeds immediately after chlorination of the methyl group of 3-picoline, resulting in the production of 3-TF. In the next step, further nuclear chlorination of the pyridine ring is performed in the empty phase to give 2,5-CTF as the major product, which can be subsequently converted to 2,3,5-DCTF. At the same time, 2-chloro-3-(trifluoromethyl)pyridine (2,3-CTF), which can be used to produce several commercial products, as discussed in sections 3 and 4, is also obtained as a minor product.
Similar reaction conditions can be applied to 2- or 4-picoline; representative products and yields are summarized in Table 1.
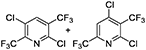
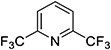
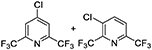
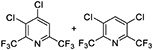
For lutidines, the reaction proceeds under similar conditions, but the reaction temperature needs to be higher than that for picolines. Several novel compounds with two trifluoromethyl groups, such as chloro-bis(trifluoromethyl)pyridine, can be synthesized in 60 to 80% yield (Table 2).
Table 2. Substrate scope of various lutidines.
a)
| Substrates and reaction temp. (°C) |
Products and yields (GC PA%) |
| CFB phase |
Empty phase |
BTF type |
CBTF type |
DCBTF type |
| 2,4-Lutidine |
|
|
|
| 420 |
420 |
5.8 |
78.8 |
13.0 |
| 2,5-Lutidine |

|

|
|
| 420 |
460 |
14.1 |
59.0 |
18.6 |
| 2,6-Lutidine |
|
|
|
| 440 |
N/A |
69.6 |
0.0 |
0.0 |
| 420 |
520 |
2.5 |
45.6 |
31.4 |
| 3,4-Lutidine |
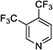
|
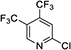
|

|
| 420 |
400 |
9.0 |
60.0 |
16.0 |
| 3,5-Lutidine |
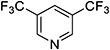
|
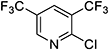
|
|
| 360 |
N/A |
89.3 |
10.7 |
0.0 |
| 380 |
440 |
14.4 |
62.2 |
21.4 |
a)Abbreviations: CFB, catalyst fluidized bed; PA%, peak area percent; BTF, bis(trifluoromethyl)pyridine; CBTF, chloro-bis(trifluoromethyl)pyridine; DCBTF, dichloro-bis(trifluoromethyl)pyridine; N/A, data not available.
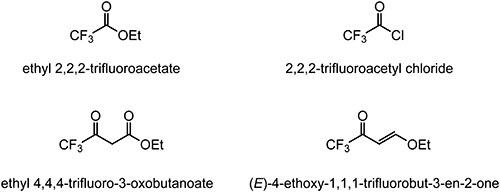
A number of cyclocondensation reactions for the synthesis of TFMP derivatives have been reported. The most commonly used trifluoromethyl-containing building blocks are ethyl 2,2,2-trifluoroacetate, 2,2,2-trifluoroacetyl chloride, ethyl 4,4,4-trifluoro-3-oxobutanoate, and (E)-4-ethoxy-1,1,1-trifluorobut-3-en-2-one (Fig. 4).
3. Agrochemicals
3.1. TFMP derivatives as agrochemicals
The ISO common names for 22 agrochemicals containing a TFMP moiety, listed in the Compendium of Pesticide Common Names (http://www.alanwood.net/pesticides/index.html), are shown in Table 3. Prior to 1990, all compounds except dithiopyr employed 3- or 5-trifluoromethyl-substituted pyridines as a partial structure. These compounds are synthesized from 2,5-CTF or 2,3,5-DCTF derived from 3-picoline. However, since 1990, other substitution patterns, mainly 6-trifluoromethyl-substituted pyridine derivatives, have increased. A 4-trifluoromethyl-substituted pyridine moiety is adopted in relatively few agrochemicals, and only flonicamid and pyroxsulam have been commercialized.
Table 3. Trifluoromethylpyridine containing agrochemicals.
a)

|
| No. |
ISO common nameb) |
CF3 position |
Indication |
CAS No. |
Year of introductionc) |
| 1 |
Fluazifop |
β (5) |
H |
69335-91-7 |
1982 (racemic) |
| 2 |
Fluazifop-P |
β (5) |
H |
83066-88-0 |
1987d) |
| 3 |
Haloxyfop |
β (5) |
H |
69806-34-4 |
1986 (racemic) |
| 4 |
Haloxyfop-P |
β (5) |
H |
95977-29-0 |
1993e) |
| 5 |
Chlorfluazuron |
β (5) |
I |
71422-67-8 |
1989 |
| 6 |
Fluazinam |
β (5) |
F |
79622-59-6 |
1988 |
| 7 |
Dithiopyr |
α (6) |
H |
97886-45-8 |
1990 |
| 8 |
Flazasulfuron |
β (3) |
H |
104040-78-0 |
1989 |
| 9 |
Picoxystrobin |
α (6) |
F |
117428-22-5 |
2001 |
| 10 |
Thiazopyr |
α (6) |
H |
117718-60-2 |
1997 |
| 11 |
Flupyrsulfuron |
α (6) |
H |
150315-10-9 |
1997 |
| 12 |
Flonicamid |
γ (4) |
I |
158062-67-0 |
2003 |
| 13 |
Pyridalyl |
β (5) |
I |
179101-81-6 |
2004 |
| 14 |
Fluopicolide |
β (5) |
F |
239110-15-7 |
2006 |
| 15 |
Bicyclopyrone |
α (6) |
H |
352010-68-5 |
2015 |
| 16 |
Pyroxsulam |
γ (4) |
H |
422556-08-9 |
2007 |
| 17 |
Fluopyram |
β (5) |
F |
658066-35-4 |
2012 |
| 18 |
Sulfoxaflor |
α (6) |
I |
946578-00-3 |
2012 |
| 19 |
Fluazaindolizine |
β f) |
N |
1254304-22-7 |
N/A |
| 20 |
Fluopimomide |
β (5) |
F |
1309859-39-9 |
2018g) |
| 21 |
Acynonapyr |
β (5) |
I |
1332838-17-1 |
2020h) |
| 22 |
Cyclobutrifluram |
α (2) |
N |
1460292-16-3 |
N/A |
a)Abbreviations: H, herbicide; I, insecticide; F, fungicide; N, nematicide; N/A, data not available. b)Compendium of Pesticide Common Names. c)PhillipsMcDougall: Product Directory 2016 Market. d)Fluazifop-P-butyl. e)Haloxyfop-P-methyl, Pesticide Properties DataBase (http://sitem.herts.ac.uk/aeru/ppdb/). f)6-(Trifluoromethyl)imidazo[1,2-a]pyridine. g)2019 China Pesticide Suppliers Guide. h)Launched in Japan (https://www.nippon-soda.co.jp/nougyo/news/pdf/20201014_1.pdf).
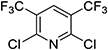
Fluazifop-butyl, the first herbicide incorporating a TFMP substructure, was commercialized by Ishihara Sangyo Kaisha, Ltd. (ISK), in 1982.19,20) Fluazifop-butyl acts as an acetyl-CoA carboxylase (ACCase) inhibitor.21) Translocation with foliar application and herbicidal activities were improved by introducing a TFMP moiety into the lead compound, such as nitrofen. These favorable features of the pyridine derivative are far superior to the corresponding benzene analogue and showed excellent herbicidal activity on perennial grass weeds.22) As shown in Scheme 5, 2,5-CTF was employed as a key intermediate for the synthesis. Fluazifop-butyl is known to have an asymmetric center on the propionate moiety, with only the (R)-enantiomer being active. After the commercialization of fluazifop-butyl (racemate), a stereoselective synthetic process for optically active fluazifop-butyl was established, and it is now marketed as fluazifop-P-butyl.
Haloxyfop-methyl was developed by Dow Chemical for post-emergence grass control in many dicotyledonous crops.23) Like fluazifop-butyl, haloxyfop-methyl targets ACCase enzyme.24) The chemical structures of these two compounds are very close to each other, whereas 2,3,5-DCTF is utilized for the preparation of haloxyfop-methyl (Fig. 5). The herbicidal spectrum of haloxyfop-methyl overlaps that of fluazifop-butyl but exhibits longer residual soil activity.25)
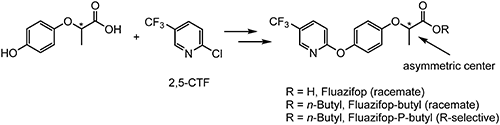
Flazasulfuron, which was first reported by ISK in 1986,26) is a sulfonylurea-type acetolactate synthase (ALS)-inhibiting herbicide that is selective on turf, sugarcane, and perennial crops such as citrus and grape.22) In the process of evaluating physicochemical and biological properties, it was found that flazasulfuron was readily decomposed and/or metabolized under various conditions through an intramolecular nucleophilic aromatic substitution reaction triggered by the large electronegativity of the trifluoromethyl moiety on the pyridine ring.27) This observation could be a breakthrough to overcome the carry-over problem with sulfonylurea herbicides and led to the invention of nicosulfuron. 2,3-CTF is employed as a key component in the preparation of flazasulfuron, as shown in Scheme 6.
Dithiopyr, discovered by the Monsanto Company,28) is a pre-emergence and early post-emergence herbicide that is used primarily in turf and ornamentals. Thiazopyr29) is a pre-emergence herbicide and has long residual control of annual grass weeds. According to the Herbicide Resistance Action Committee (HRAC) classification,30) the mode of action of dithiopyr and thiazopyr is classified as Group 3, which inhibits root growth by blocking cell division as a microtubulin assembly inhibitor.31) The summarized synthetic pathways are shown in Scheme 7. The synthesis of both compounds starts with a cyclocondensation reaction of 3-methylbutanal and ethyl 4,4,4-trifluoro-3-oxobutanoate as a key trifluoromethyl-containing building block to give tetrahydro-2H-pyran (1). Then 1 is converted to compound 2 as a common intermediate of dithiopyr and thiazopyr.32)

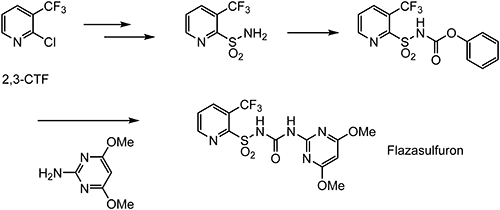
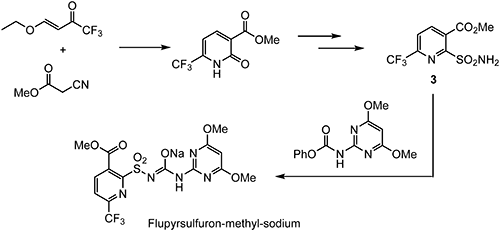
Flupyrsulfuron-methyl-sodium is an ALS-inhibiting cereal herbicide discovered by DuPont.33) Selective control of Alopecurus myosuroides, Apera spica-venti, and a wide range of broadleaf weeds is obtained through post-emergence application.34) The chemical structure of flupyrsulfuron-methyl-sodium and a typical preparation method of 2-sulfamoyl-6-(trifluoromethyl)nicotinic acid methyl ester (3) as a key intermediate are shown in Scheme 8.35) Crop selectivity derives from the rapid metabolism degradation of flupyrsulfuron-methyl-sodium in wheat and very slow metabolism in sensitive weeds.36) Since the electron-deficient pyridine moiety is substituted by strong electron-withdrawing groups, such as trifluoromethyl and methoxycarbonyl moieties, it is susceptible to the aromatic nucleophilic substitution and degrades quickly in soil, water, and sediment.37)
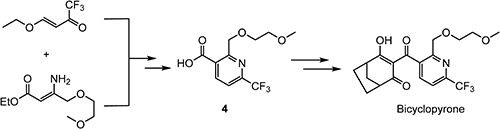
Bicyclopyrone38) was discovered as a 4-hydroxyphenylpyruvate dioxygenase (HPPD)-inhibiting herbicide for use in corn and launched by Syngenta in 2015. Intermediate 4 was prepared by a cyclization reaction using (E)-4-ethoxy-1,1,1-trifluorobut-3-en-2-one as shown in Scheme 9.39) The pyridyl nitrogen in bicyclopyrone has the effect of reducing hydrophobicity and increasing acidity. It can be inferred that these characteristics may have led to improvement in the bioavailability of pre-emergent applications, compared with the corresponding phenyl analogue.40)
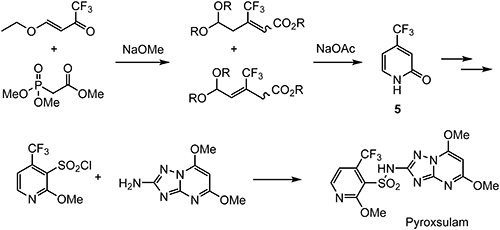
Pyroxsulam,41) discovered by Dow AgroSciences LLC, is an ALS-inhibiting herbicide with the characteristic triazolopyrimidine and 2-methoxy-4-(trifluoromethyl)pyridine substructures.42,43) It was developed for the control of key weeds in cereal crops such as wheat. High herbicidal activity on grass species was achieved with 2-methoxy-4-(trifluoromethyl)phenyl analogues, but these compounds were found to cause significant injury to wheat, unlike pyridine analogues.44) The selectivity of pyroxsulam to wheat relatives is connected primarily with differences in the rate of metabolism.45) As shown in Scheme 10, the pyridone intermediate 5 is synthesized by the Horner–Wadsworth–Emmons reaction using (E)-4-ethoxy-1,1,1-trifluorobut-3-en-2-one as a trifluoromethyl-containing building block.46)
3.3. Insecticide
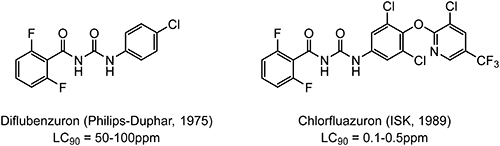
Chlorfluazuron,47) discovered by ISK, has a typical benzoylurea-type structure, which is well known as an insect growth regulator (IGR). Chlorfluazuron exhibits excellent insecticidal activity against specific target insects, such as Lepidoptera, Diptera, and Orthoptera, at their larval stages through the chitin biosynthesis-inhibition mechanism. As shown in Fig. 6, chlorfluazuron showed higher insecticidal activity against the common cutworm as compared with the lead compound diflubenzuron.22) 2,3,5-DCTF was employed as a starting raw material for the synthesis of chlorfluazuron, as shown in Scheme 11.
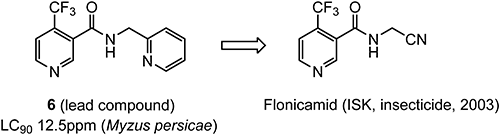
Flonicamid was discovered by ISK as a novel insecticide to control aphids.48) The initial screening of various TFMP analogues revealed that some nicotinamide derivatives showed interesting activity against aphids, as shown in Fig. 7.49) Structural optimization using compound 6 as a lead compound led to the discovery of flonicamid, which has a unique 4-trifluoromethyl-substituted pyridine moiety. Flonicamid is a chordotonal organ modulator and is classified as Group 29 according to the Insecticide Resistance Action Committee (IRAC) mode of action classification.50) The synthesis route of the 4-(trifluoromethyl)nicotinic acid is shown in Scheme 12.51)
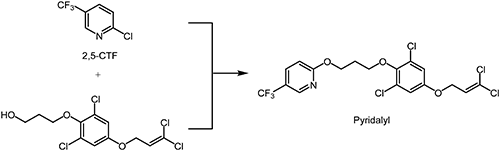
Pyridalyl was discovered and reported by Sumitomo Chemical Co., Ltd.52,53) It has a 5-(trifluoromethyl)pyridine moiety as a key substructure in the molecule. According to the IRAC, the mode of action of pyridalyl is classified as unknown (UN).50,54) After structural optimization, the insecticidal activity of TFMP derivatives on lepidopterous pests was found to be superior to that of the corresponding phenyl analogues. The chemical structure and synthetic route of pyridalyl are shown in Scheme 13.55)
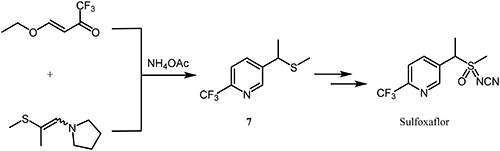
Sulfoxaflor,56,57) commercialized by Dow AgroSciences LLC in 2012, is an insecticide targeting sap-feeding pests. The sulfoxyimine functional group is rare and characteristic as a pesticide. The IRAC has classified sulfoxaflor as the only insecticide in Group 4, Subgroup 4C.50) At the beginning of the novel sulfoxyimine analogue research, phenyl derivatives that exhibited weak fungicidal activity were explored. However, when the phenyl moiety was replaced with a hetero-aromatic ring such as pyridine, insecticidal activity was observed. As a result of further structural optimization, it was found that the 6-(trifluoromethyl)pyridine moiety was the optimal substructure, showing excellent insecticidal activity.58,59) A practical preparation route of sulfoxaflor is shown in Scheme 14. A critical pyridine sulfide intermediate, 7, is prepared via an enamine-mediated cyclocondensation reaction by using (E)-4-ethoxy-1,1,1-trifluorobut-3-en-2-one as a trifluoromethyl-containing building block.60)
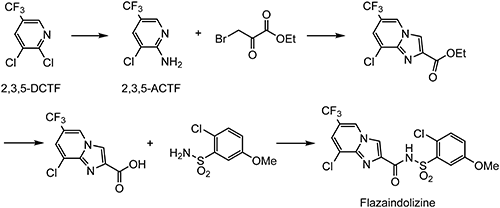
N-phenylsulfonylimidazopyridine-2-carboxamide derivatives were found via high-throughput screening of DuPont’s internal compound libraries against a root-knot nematode. Fluazaindolizine61) was selected as the optimal compound after structural optimization aiming at both the reduction of phytotoxicity and the improvement of biological activity. The trifluoromethyl group at the 6-position of the imidazopyridine moiety is particularly important for nematicidal activity. Therefore, fluazaindolizine shows activity superior to the corresponding unsubstituted or halogen-substituted imidazopyridine derivatives.62) According to the IRAC classification, the mode of action of fluazaindolizine is UN, but it seems to be novel, because there is no activity against the target sites of current commercial nematicides. 2-Amino-3-chloro-5-(trifluoromethyl)pyridine (2,3,5-ACTF), which can be prepared from 2,3,5-DCTF, was employed as a starting material, as shown in Scheme 15.
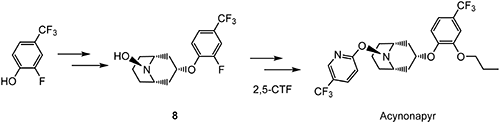
Acynonapyr63) is a new acaricide discovered by Nippon Soda and is active on spider mites of Tetranychus and Panonychus. Acynonapyr has a characteristic chemical structure in which azabicyclo[3.3.1]nonane core 864) and the 5-(trifluoromethyl)pyridine ring are connected via an ether bond, as shown in Scheme 16. The mode of action of acynonapyr has not yet been classified but is presumed to be disturbance of the neurotransmitter system by acting on inhibitory glutamate receptors.65)
The first patent claimed for cyclobutrifluram was published in 2013 by Syngenta.66) Cyclobutrifluram has been reported as a nematicide and is characterized by a novel carboxamide structure that has a cis-substituted four-membered ring comprised of a specific absolute configuration at each of two positions.67,68) Although there is no information on the mechanism of action, its chemical structure is reminiscent of fluopyram, with the same nematicidal effect. 2-(Trifluoromethyl)nicotinic acid is expected to be a critical raw material for the synthesis of cyclobutrifluram. Several preparation methods of 2-(trifluoromethyl)nicotinic acid have been reported in previous literature.69–71) One of the representative synthetic methods is a cyclocondensation reaction starting from compound 9, derived from Vilsmeier salt, as shown in Scheme 17.72) Ethyl 4,4,4-trifluoroacetoacetate can be condensed with compound 9 to form compound 10, followed by intramolecular cyclization to obtain 2-(trifluoromethyl)nicotinic acid.

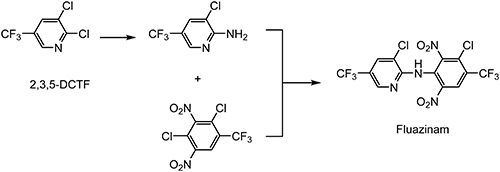
Fluazinam, discovered by ISK,73) is a broad-spectrum fungicide, and its mode of action is the uncoupling of oxidative phosphorylation, classified as Fungicide Resistance Action Committee (FRAC) code 29.74) Substituent effects on the pyridine ring were examined using the fungicidal activity against Botrytis cinerea. It was found that the trifluoromethyl-substituted pyridine derivative showed higher fungicidal activity than the corresponding chloro-, nitro-, and cyano-substituted derivatives.75) 2,3,5-DCTF is utilized in the synthesis of fluazinam, as shown in Scheme 18.
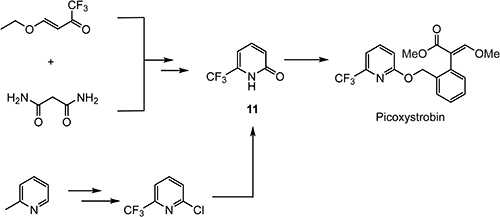
Picoxystrobin76) was discovered as a strobilurin fungicide and developed by Syngenta for the European market. After a while, it was passed to DuPont and developed for wheat in North America and Asia and for soybean crops in South America in the mid 2000s. One structural characteristic of picoxystrobin is a pyridine ring in which the 6-position is substituted with a trifluoromethyl group. Two procedures for the preparation of 6-(trifluoromethyl)pyridin-2(1H)-one (11) have been reported: a cyclocondensation method starting from (E)-4-ethoxy-1,1,1-trifluorobut-3-en-2-one and direct fluorination of 2-picoline through a chlorine/fluorine exchange reaction, as shown in Scheme 19.77–79)
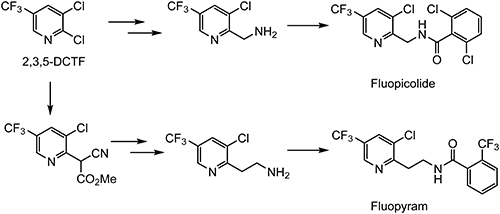
Bayer’s chemists focused on the 3-chloro-5-(trifluoromethyl)pyridine residue based on the “Agrophore Approach”80) and discovered two commercial fungicides.81) It is quite interesting that the same fungicidal mechanism is inferred from the similarity of both chemical structures, but they are different according to the FRAC classification. Fluopicolide induces the delocalization of spectrin-like proteins in oomycete fungi, which is a new mode of action classified as FRAC code 43. On the other hand, fluopyram is a succinate dehydrogenase inhibitor (SDHI, FRAC code 7) fungicide that is effective on ascomycete pathogens and plant-parasitic nematodes.82) Chemical structures and outlines of the synthesis route for both fluopicolide83) and fluopyram84) are shown in Scheme 20.
Fluopimomide85) was developed by Shandong Sino-Agri United Biotechnology Co., Ltd., and approved in China in 2017. As shown in Fig. 8, it has a chemical structure very similar to the preceding fungicide, fluopicolide, but its EC50 against Pseudoperonospora cubensis is relatively low, and the observed downward transportation is different from that of fluopicolide.86) A nematicidal effect has also been reported for fluopimomide.87) 5-(Trifluoromethyl)pyridine is incorporated as a substructure as well as fluopicolide, whereas the substituents on the benzene ring are characteristic. According to the FRAC classification, both compounds are reported to have the same mode of action and are classified as code 43.
4. Pharmaceuticals
4.1. Overview of TFMP derivatives in the pharmaceuticals
In 2010, it was estimated that up to 20% of pharmaceuticals prescribed or administered in the clinic contained a fluorine atom.88) More recently, however, this has increased to over 40%. It is noteworthy that 19.2% of those fluorinated drugs contain a trifluoromethyl group.89)
Five pharmaceuticals (Table 4) containing a TFMP moiety as a partial structure have been approved and commercialized so far. Furthermore, since various drug candidates containing a TFMP moiety are currently undergoing clinical trials, new drugs are expected to be introduced to the market in the near future. Representative examples of drugs under development and their common names are listed in Table 5.90)
Table 4. Approved drugs.
| No. |
Development code |
Common namea) |
CF3 position |
Efficacy disease |
CAS No. |
Approval dateb,c) |
| 1 |
PNU-140690 |
Tipranavir |
β (5) |
Antivirals, HIV-1 infection |
174484-41-4 |
2005/6/22 FDA |
| 2005/10/25 EMA |
| 2 |
AG-221 CC-90007 |
Enasidenib |
α (2 and 6) |
Antineoplastic, Acute myeloid Leukemia |
1446502-11-9 |
2017/8/1 FDA |
| 3 |
ARN-509 JNJ-927 |
Apalutamide |
β (5) |
Antineoplastic, Prostate Cancer |
956104-40-8 |
2018/2/14 FDA |
| 2019/1/14 EMA |
| 2019/3/26 PMDA |
| 4 |
MK-1439 |
Doravirine |
γ (4) |
Antivirals, HIV-1 infection |
1338225-97-0 |
2018/8/30 FDA |
| 2018/11/22 EMA |
| 2020/1/14 PMDA |
| 5 |
PLX3397 |
Pexidartinib |
α (6) |
Antineoplastic, Giant cell tumor of tendon sheath |
1029044-16-3 |
2019/9/5 FDA |
a)International Nonproprietary Name (https://druginfo.nlm.nih.gov/drugportal/); b)As of Dec. 2020. c)Abbreviations: FDA, Food and Drug Administration; EMA, European Medicines Agency; PMDA, Pharmaceuticals and Medical Devices Agency.
Table 5. Drugs in development.
| No. |
Development code |
Common name |
CF3 position |
Efficacy disease |
CAS No. |
Development stageb) |
| 1 |
CVL-751 PF-06649751 |
Tavapadon |
β (3) |
Antiparkinsonians |
1643489-24-0 |
Phase III (USA) |
| 2 |
CDZ173 |
Leniolisiba) |
β (5) |
Immunodeficiency disorders |
1354690-24-6 |
Phase II/III (USA,EU) |
| 3 |
CORT125134 |
Relacorilanta) |
γ (4) |
Prostate cancer Cushing syndrome |
1496510-51-0 |
Phase II/III (USA, EU) |
| 4 |
PQR309 |
Bimiralisiba) |
γ (4) |
Head and neck squamous cell cancer (HNSCC) |
1225037-39-7 |
Phase II (USA) |
| 5 |
QBW-251 |
Icenticaftora) |
β (5) |
Chronic obstructive pulmonary disease (COPD) |
1334546-77-8 |
Phase II (USA, EU, JP) |
| 6 |
SL-801 CBS9106 |
Felezonexor |
β (5) |
Antineoplastic |
1076235-04-5 |
Phase I (USA) |
| 7 |
LXH254 |
Naporafenib |
α (2) |
Antineoplastic |
1800398-38-2 |
Phase I (USA, EU, JP) |
a)International Nonproprietary Name (https://druginfo.nlm.nih.gov/drugportal/); b)As of Dec. 2020.
In 2005, Boehringer Ingelheim succeeded in developing and commercializing tipranavir,91) which was discovered by Pharmacia & Upjohn (currently Pfizer, Inc.),92) as a non-peptide anti-human immunodeficiency virus (HIV) drug.
In the course of structural optimization, for all synthesized compounds, not only the HIV protease binding activity but also the relationship between pharmacokinetic parameters, such as in vitro antiviral activity and half-life in blood, was comprehensively investigated. Tipranavir with a TFMP moiety showed about tenfold higher antiviral activity than the corresponding phenyl analogue.
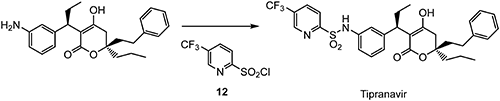
The first synthesis93) of tipranavir using a chiral auxiliary was reported in 1997, but later, a practical asymmetric synthesis was also reported. The introduction of 5-(trifluoromethyl)pyridine-2-sulfonyl chloride (12) prepared from 2,5-CTF is achieved in the final synthesis step, as shown in Scheme 21.94)
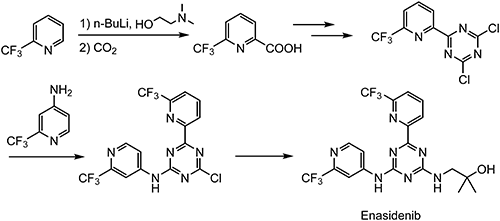
Enasidenib is an oral isocitrate dehydrogenase-2 (IDH2) inhibitor developed by the Celgene Corp. under a global, exclusive license from Agios Pharmaceuticals, Inc.95)
In 2017, the United States Food and Drug Administration (FDA) approved enasidenib for adults with relapsed and refractory acute myelogenous leukemia (AML) with an IDH2 mutation.
The synthesis of enasidenib consists of six steps, depicted in Scheme 22.96) One structural characteristic of enasidenib is that two different α-TFMPs are introduced into both ends of the molecule.
Several synthetic methods of 2-(trifluoromethyl)pyridine (13) and 2-(trifluoromethyl)pyridin-4-amine (14), shown in Scheme 23, were reported previously.97,98)

Scheme 23. Synthesis of TFMP intermediates.
Apalutamide,99) developed by Janssen Pharmaceuticals, Inc., is a second-generation nonsteroidal androgen receptor (AR) antagonist with a strong binding affinity to AR. In 2018, it was approved by the FDA for the treatment of non-metastatic castration-resistant prostate cancer (nmCRPC). As shown in Fig. 9, the chemical structure of apalutamide is very similar to that of enzalutamide (FDA approved in 2012) with two minor modifications: (a) a gem-dimethyl group on the imidazoline ring of enzalutamide is bonded at each end to form a four-membered cyclic structure, and (b) the benzene ring of enzalutamide is replaced with a pyridine ring. Apalutamide possesses in vitro activity comparable to that of enzalutamide but with greater anti-tumor efficacy in castration-resistant prostate cancer (CRPC) xenograft models and lower potential to cause seizures as an adverse effect in the central nervous system.100)
A detailed review of the development of synthetic approaches to apalutamide has been published.101) An example of the synthetic route is shown in Scheme 24, in which 2,3-CTF is employed as a key intermediate.
Doravirine,102) developed by the Merck Sharp & Dohme Corp., is a new non-nucleoside reverse transcriptase inhibitor (NNRTI), and it was approved by the FDA in 2018 for the treatment of HIV infection in adult patients. Similar to other NNRTIs, doravirine exerts its antiviral effect through a noncompetitive inhibition of the HIV-1 reverse transcriptase but has a more favorable drug interaction profile compared with earlier NNRTIs, as it neither inhibits nor induces the cytochrome P450 3A4 (CYP3A4) enzyme.103)
In structural optimization, a conversion of the methyl group on the pyridone ring to a trifluoromethyl group was found to improve plasma stability and enzyme inhibition.104,105)
A robust kilo-scale synthesis route of doravirine, shown in Scheme 25, has been reported,106) but details of the preparation method of 2-chloro-3-fluoro-4-(trifluoromethyl)pyridine (15) have not been described in the literature.
As an alternative synthesis route for the key pyridine intermediate of doravirine, a flow reactor makes it possible to use (E)-4-ethoxy-1,1,1-trifluorobut-3-en-2-one as a starting raw material instead of 15, as shown in Scheme 26.107)
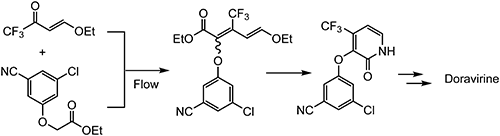
Scheme 26. Alternative synthesis route of doravirine.
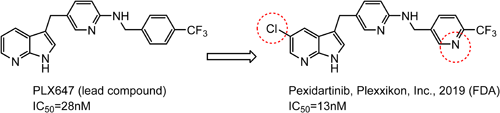
Pexidartinib,108) discovered by Plexxikon, Inc., is an orally active and potent colony-stimulating factor-1 receptor (CSF-1R) kinase inhibitor (IC50 value 13 nM).109)
In 2019, the FDA approved Daiichi Sankyo’s pexidartinib capsules for the treatment of adult patients with symptomatic tenosynovial giant cell tumors (TGC Ts) associated with severe morbidity or functional limitations and not amenable to improvement with surgery.
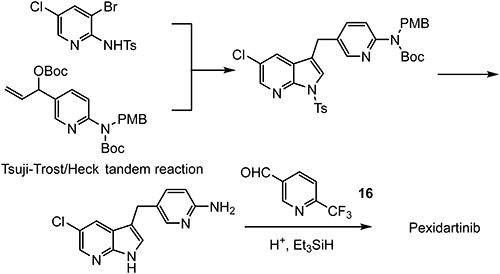
As shown in Fig. 10, pexidartinib is the compound in which a chlorine atom is introduced at the 5-position of the 1H-pyrrolo[2,3-b]pyridine moiety of PLX647 (lead compound), and the benzene ring is replaced with a pyridine. The CSF-1R kinase inhibitory activity is about twofold higher, and the nitrogen atom of the pyridine ring contributes to the stabilization of the CSF-1R kinase conformation.
A novel synthetic route (Scheme 27) was designed and demonstrated.110) 6-(Trifluoromethyl)nicotinaldehyde (16) was used as a key intermediate for the synthesis of pexidartinib.
Compound 16 is synthesized by a cyclocondensation reaction using (E)-4-ethoxy-1,1,1-trifluorobut-3-en-2-one, as shown in Scheme 28.111–113)
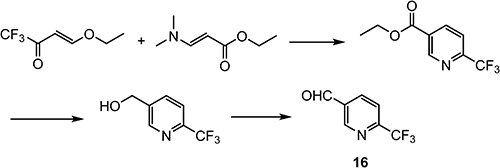
Scheme 28. Synthesis of 6-(trifluoromethyl)nicotinaldehyde (16).
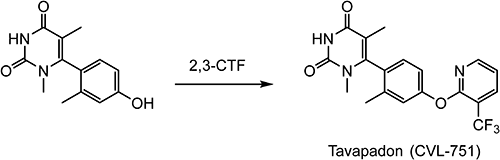
Tavapadon (CVL-751),114) discovered by Pfizer, Inc., is a potent partial agonist at the D1/D5 receptors and it is being developed by Cerevel Therapeutics as a treatment for Parkinson’s disease.115) 2,3-CTF is employed as a key intermediate for the preparation of CVL-751 as shown in Scheme 29.
Leniolisib (CDZ173),116) discovered by Novartis AG, is currently in clinical studies by Pharming in patients suffering from activated PI3Kδ syndrome (APDS)/p110 delta activating mutation causing senescent T cells, lymphadenopathy, and immunodeficiency (PASLI). In structural optimization, conversion of the methyl group on the pyridine moiety to a trifluoromethyl group was found to increase PI3Kδ potency by a factor of five. Leniolisib was found to have the optimal hydrophilic property to ensure good solubility and metabolic stability.117)
2,3-CTF is employed as a key intermediate for the preparation of leniolisib, as shown in Scheme 30.
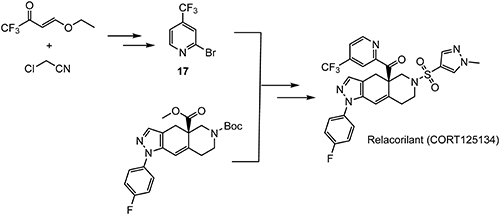
Relacorilant (CORT125134)118) is being developed by Corcept Therapeutics, Inc. It is an orally active, high-affinity, selective antagonist of the glucocorticoid receptor that may benefit from the modulation of cortisol activity. In structural optimization, the introduction of a trifluoromethyl group to the 4-position on the pyridyl moiety was found to increase HepG2 tyrosine amino transferase assay potency by a factor of four. Relacorilant is currently being evaluated in a phase II clinical study in patients with Cushing’s syndrome.119)
2-Bromo-4-(trifluoromethyl)pyridine (17) prepared from (E)-4-ethoxy-1,1,1-trifluorobut-3-en-2-one is employed as a key intermediate for the preparation of relacorilant as shown in Scheme 31.120)
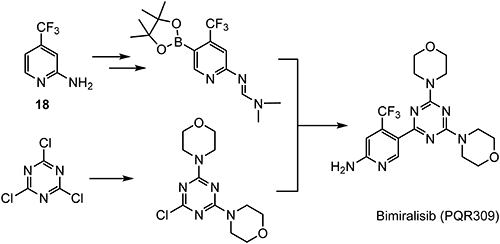
Bimiralisib (PQR309)121) is being developed by PIQUR Therapeutics AG. It has been selected after structural optimization as the best phosphoinositide 3-kinase(PI3K) inhibitor with a balanced targeting of the mammalian target of the rapamycin (mTOR) kinase.122) Bimiralisib evolved as the lead compound of a series of dimorpholinotriazine-based compounds and is currently being tested in phase II clinical trials.
2-Amino-4-(trifluoromethyl)pyridine (18), which can be prepared from 2,4-CTF, is employed as a key intermediate for the preparation of bimiralisib, as shown in Scheme 32.123)
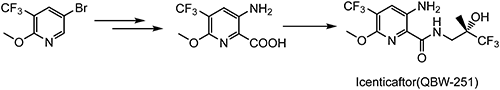
Icenticaftor (QBW-251), which is a potentiator of the cystic fibrosis membrane conductance regulator (CFTR)124) used for the treatment of chronic obstructive pulmonary disease (COPD), is being developed by Novartis AG.
5-Bromo-2-methoxy-3-(trifluoromethyl)pyridine (the same intermediate of leniolisib), which can be prepared from 2,3-CTF, is employed as a key intermediate for the preparation of icenticaftor, as shown in Scheme 33.125)
Chromosome region maintenance 1 (CRM1) plays an important role in the nuclear export of cargo proteins bearing nuclear exporting signal sequences. Felezonexor (CBS9106),126) which was discovered by the CanBas Co., Ltd., is a novel reversible CRM1 inhibitor and a promising clinical candidate developed by Stemline Therapeutics, Inc.127)
2,6-Dichloro-3-(trifluoromethyl)pyridine (2,6,3-DCTF) is employed as a starting material for the preparation of felezonexor, as shown in Scheme 34.
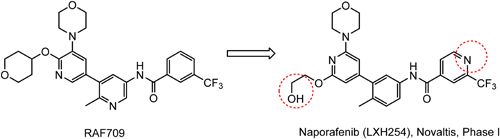
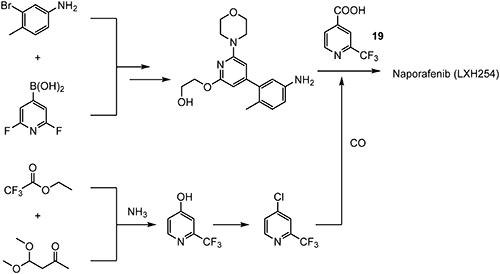
RAF709128) was a potent, selective, efficacious rapidly accelerated fibrosarcoma (RAF) kinase inhibitor and well tolerated in preclinical models, but the high human intrinsic clearance precluded further development and prompted further investigation of close analogues. In further structural optimization studies, the mitigation of human intrinsic clearance and time-dependent inhibition led to the discovery of naporafenib (LXH254), as shown in Fig. 11.129,130) Naporafenib was advanced to human studies and is currently being assessed in phase I trials by the Novartis Pharmaceuticals Corporation.
An optimized synthetic route for naporafenib, which was used to supply larger quantities of material for in vivo studies, is outlined in Scheme 35. 2-(Trifluoromethyl)isonicotinic acid (19) is introduced in the last step of the synthesis, as shown in Scheme 35.131,132) For the synthesis of compound 19, ethyl 2,2,2-trifluoroacetate is used as a key precursor.
5. Animal health products
Two animal health products (Table 6) containing a TFMP moiety have been approved and commercialized.
Table 6. Approved animal health products.
| No. |
Development code |
Common name |
CF3 position |
Efficacy disease |
CAS No. |
Approved datea) |
| 1 |
VB0PV6I7L6 |
Fluazuronb) |
β (5) |
Tick control in beef cattle |
86811-58-7 |
1994 |
| 2 |
IS-741 IKV-741 |
Fuzapladib |
β (5) |
Acute pancreatitis drug for dogs |
351999-87-6c) |
2018 |
a)As of Dec. 2020; b)International Nonproprietary Name (https://druginfo.nlm.nih.gov/drugportal/); c)Fuzapladib sodium hydrate.
Fluazuron, a benzoyl-phenylurea derivative, is a well-known chemical class of chitin biosynthesis inhibitors and was developed exclusively for veterinary use by Ciba-Geigy.133) It shows good activity against ticks and mites of livestock and pets but none at all against insects. Fluazuron was approved as an animal health product in 1994.134) TFMP is incorporated as a substructure in fluazuron, as shown in Fig. 12.
Fuzapladib sodium hydrate (IKV-741, IS-741) was discovered and developed by ISK as an acute pancreatitis drug for dogs.
IS-741 had been a clinical candidate for patients with acute pancreatitis,135) but further development was cancelled in phase II. Later, this compound continued to be developed as a veterinary medicine and was approved as fuzapladib sodium hydrate in 2018.136)
Fuzapladib sodium hydrate is synthesized from 2,5-CTF as a starting material, as shown in Scheme 36.137)
Conclusion
Despite not occurring in nature,138) fluorine derivatives have attracted attention as a rich source of bioactive compounds. Herein, we have discussed the synthesis and applications of TFMP derivatives. In the more than 40 years since the first TFMP-based agrochemical was reported, research and development of TFMP derivatives has continued unabated, and many new bioactive molecules have been discovered. Although TFMP derivatives were first applied in the agrochemical industry, their use in the pharmaceutical industry is increasing. Early structural design introduced the TFMP motif into a compound to manipulate its physicochemical properties and obtain better biological activity. There are also reports that TFMP itself has various biological activities. For instance, 2-amino-3-chloro-5-(trifluoromethyl)pyridine is expected to show anti-tumor activity,139) and 3-chloro-5-(trifluoromethyl)pyridine-2-carboxylic acid, a metabolite of the fungicide fluopyram, is phytotoxic.140) More agrochemicals and pharmaceuticals containing TFMP are expected to be introduced to the market in the near future. For discovery chemists, confirming the biological activity of TFMP derivatives during the structural optimization stage of the design process may open several useful avenues for future research.
References
- 1) Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata: Current Contributions of Organofluorine Compounds to the Agrochemical Industry. iScience 23, 101467 (2020).
- 2) A. Bondi: van der Waals Volumes and Radii. J. Phys. Chem. 68, 441–451 (1964).
- 3) A. L. Allred: ELECTRONEGATIVITY VALUES FROM THERMOCHEMICAL DATA. J. Inorg. Nucl. Chem. 17, 215–221 (1961).
- 4) C. Hansch, A. Leo and R. W. Taft: A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev. 91, 165–195 (1991).
- 5) J. E. Huheey: The Electronegativity of Groups. J. Phys. Chem. 69, 3284–3291 (1965).
- 6) F. Swarts: Fluorine derivatives of toluene. Bull. Acad. Roy. Belg. 35, 375–420 (1898).
- 7) P. Osswald, F. Müller and F. Steinhäuser: (IG Farbenindustrie AG): German Pat. Appl. DE 575593 (1933).
- 8) E. T. McBee, H. B. Hass and E. M. Hodnett: FLUORINATED HETEROCYCLIC COMPOUNDS methylpyridines. Ind. Eng. Chem. Res. 39, 389–391 (1947).
- 9) https://www.cas.org/sites/default/files/documents/registry.pdf (accessed Feb. 4, 2021).
- 10) https://www.stn-international.com/sites/default/files/stn/dbss/CAPLUS.pdf (accessed Apr. 28, 2021).
- 11) D. G. Kananovich: Copper-Catalyzed Trifluoromethylation Reactions. In “Copper Catalysis in Organic Synthesis,” eds. by G. Anilkumar and S. Saranya, Wiley-VCH, Weinheim, pp. 367–393, (2020).
- 12) H. Li, K. Jin, Y. Wu, Y. Zhao and H. Jin: (Lianyungang Avilive Chemical Co., Ltd. and Zhejiang Avilive Chemical Co., Ltd.): Chinese Pat. Appl. CN 105439943 (2016).
- 13) J. A. Werner, C. A. Wilson and C. E. Mixan: (The Dow Chemical Company): US Pat. Appl. US 4331811 (1982).
- 14) X. Dai, J. Zhang and Y. Tang: (Kangpeng Chemistry Co., Ltd.): Chinese Pat. Appl. CN 101092392 (2007).
- 15) K. D. Campbell, D. A. Livingston, H. S. Wan, B. J. Schoeman, S. R. Lakso and K. M. Larson (Dow AgroSciences LLC): PCT Int. Appl. WO 2005/105747 (2005).
- 16) A. P. Fung, C. A. Wilson, G. S. Fujioka and J. A. Werner (The Dow Chemical Company): Eur. Pat. Appl. EP 0110690 (1984).
- 17) R. Nishiyama, K. Fujikawa, I. Yokomichi, Y Tsujii and S. Nishimura (Ishihara Sangyo Kaisha Ltd.): Eur. Pat. Appl. EP 042696 (1981).
- 18) R. Nishiyama: The chlorofluorination of β-picoline. Shokubai 32, 246–252 (1990) (in Japanese).
- 19) R. Nishiyama, T. Haga and N. Sakashita: (Ishihara Sangyo Kaisha Ltd.): Jpn. Kokai Tokkyo Koho JP 54119476 (1979).
- 20) T. Haga, K. Fujikawa, N. Sakashita and R. Nishiyama: Development of a new herbicide, fluazifop-butyl. Japanese J. Pestic. Sci. 12, 311–324 (1987) (in Japanese).
- 21) K. A. Walker, S. M. Ridley, T. Lewis and J. L. Harwood: Fluazifop, a grass-selective herbicide which inhibits acetyl-CoA carboxylase in sensitive plant species. Biochem. J. 254, 307–310 (1988).
- 22) T. Haga, Y. Tsujii, K. Hayashi, F. Kimura, N. Sakashita and K. Fujikawa: Trifluoromethylpyridines as Building blocks for new agrochemicals. In “Synthesis and Chemistry of Agrochemicals II,” eds. by D. R. Baker, J. G. Fenyes and W. K. Moberg, ACS Symposium series 443. ACS Washington, DC, pp. 107–119 (1991).
- 23) D. Cartwright: (Imperial Chemical Industries Limited): UK Pat. Appl. GB 2002368 (1979).
- 24) H. Zhang, Z. Yang, Y. Shen and L. Tong: Crystal Structure of the Carboxyltransferase Domain of Acetyl-Coenzyme A Carboxylase. Science 299, 2064–2067 (2003).
- 25) D. D. Buhler and O. C. Burnside: Herbicidal Activity of Fluazifop-Butyl, Haloxyfop-Methyl, and Sethoxydim in Soil. Weed Sci. 32, 824–831 (1984).
- 26) F. Kimura, T. Haga, N. Sakashita, C. Honda, K. Hayashi, T. Seki and K. Minamida (Ishihara Sangyo Kaisha Ltd.): Eur. Pat. Appl. EP 0184385 (1986).
- 27) S. Murai, T. Haga, N. Sakashita, Y. Nakamura, C. Honda, S. Honzawa, F. Kimura, Y. Tsujii and R. Nishiyama: Synthesis and Herbicidal Activity of Sulfonylureas; SL-950 and Its Related Compounds. J. Pestic. Sci. 20, 453–462 (1995).
- 28) L. F. Lee (Monsanto Company): Eur. Pat. Appl. EP 0133612 (1985).
- 29) Y. L. L. Sing and L. F. Lee (Monsanto Company): Eur. Pat. Appl. EP 0278944 (1988).
- 30) HRAC https://hracglobal.com/tools/classification-lookup (accessed Nov. 11, 2020).
- 31) B. L. Armbruster, W. T. Molin and M. W. Bugg: Effect of the Herbicide Dithiopyr on Cell Division in Wheat Root Tips. Pestic. Biochem. Physiol. 39, 110–120 (1991).
- 32) L. F. Lee, G. L. Stikes, L. Y. L. Sing, M. L. Miller, M. G. Dolson, J. E. Normansell and S. M. Auinbauh: Synthesis of a New Class of Pyridine Herbicide. Pestic. Sci. 31, 555–568 (1991).
- 33) T. A. Andrea and P. H. T. Liang (E.I. Du Pont de Nemours and Company): Eur. Pat. Appl. EP 0502740 (1992).
- 34) S. R. Teaney, L. Armstrong, K. Bentley, D. Cotterman, D. Leep, P. H. Liang, C. Powley and J. Summers: DPX.KE459. A New Sulfonylurea for Postemergence Grass and Broadleaf Weed Control in Cereals. Proc. Brighton Crop Protection Conference—Weeds 1. 49–56 (1995).
- 35) G. A. Bullock and G. C. Chiang (E.I. Du Pont de Nemours and Company): Eur. Pat. Appl. EP 0522392 (1992).
- 36) M. K. Koeppe, A. C. Barefoot, C. D. Cotterman, W. T. Zimmerman and D. C. Leep: Basis of Selectivity of the Herbicide Flupyrsulfuron-methyl in Wheat. Pestic. Biochem. Physiol. 59, 105–117 (1998).
- 37) S. K. Singles, G. M. Dean, D. M. Kirkpatrick, B. C. Mayo, A. D. Langford-Pollard, A. C. Barefoot and F. Q. Bramble Jr.: Fate and behavior of flupyrsulfuron-methyl in soil and aquatic systems. Pestic. Sci. 55, 288–300 (1999).
- 38) A. Edmunds, A. Demesmaeker, C. Lüthy and J. Schaetzer (Syngenta Participations AG): PCT Int. Appl. WO 01/94339 (2001).
- 39) D. A. Jackson, M. C. Bowden (Syngenta Participations AG): PCT Int. Appl. WO 2004/078729 (2004).
- 40) A. Burriss, A. J. F. Edmunds, D. Emery, R. G. Hall, O. Jacob and J. Schaetzer: The importance of trifluoromethyl pyridines in crop protection. Pest Manag. Sci. 74, 1228–1238 (2018).
- 41) T. C. Johnson, J. C. Vanheertum, D. G. Ouse, M. A. Pobanz, K. E. Arndt and D. K. Walker (Dow AgroSciences LLC): PCT Int. Appl. WO 02/36595 (2002).
- 42) B. M. Bell, P. E. Fanwick, P. R. Graupner and G. A. Roth: Application of the Tisler Triazolopyrimidine Cyclization to the Synthesis of a Crop Protection Agent and an Intermediate. Org. Process Res. Dev. 10, 1167–1171 (2006).
- 43) M. A. Gonzalez, D. B. Gorman, C. T. Hamilton and G. A. Roth: Process Development for the Sulfonamide Herbicide Pyroxsulam. Org. Process Res. Dev. 12, 301–303 (2008).
- 44) T. C. Johnson, R. K. Mann, P. R. Schmitzer, R. E. Gast and G. J. deBoer: “Modern Crop Protection Compounds” eds. by P. Jeschke, M. Witschel, W. Krämer and U. Schirmer, Wiley-VCH Weinheim, pp. 106–125, 2019.
- 45) G. J. deBoer, S. Thornburgh, J. Gilbert and R. E. Gast: The impact of uptake, translocation and metabolism on the differential selectivity between blackgrass and wheat for the herbicide pyroxsulam. Pest Manag. Sci. 67, 279–286 (2011).
- 46) C. T. Hamilton, M. F. Gullo, M. A. Gonzalez and D. B. Gorman: (Dow AgroSciences LLC): US Pat. Appl. US 20050288511 (2005).
- 47) R. Nishiyama, K. Fujikawa, M. Nasu, T. Toki and T. Yamamoto: (Ishihara Sangyo Kaisha, Ltd.): German Pat. Appl. DE 2818830 (1979).
- 48) T. Toki, T. Koyanagi, M. Morita, T. Yoneda, C. Kagimoto, and H. Okada (Ishihara Sangyo Kaisha, Ltd.): Eur. Pat. Appl. EP 0580374 (1994).
- 49) M. Morita, T. Yoneda and N. Akiyoshi: Research and development of novel insecticide, flonicamid. Japanese J. Pestic. Sci. 39, 127–133 (2014) (in Japanese).
- 50) IRAC https://irac-online.org/modes-of-action/ (Accessed Nov. 11, 2020).
- 51) T. Koyanagi, T. Yoneda, F. Kanamori, S. Kanbayashi, T. Tanimura and N. Horiuchi (Ishihara Sangyo Kaisha Ltd.): Eur. Pat. Appl. EP 0744400 (1996).
- 52) N. Sakamoto, S. Matsuo, M. Suzuki, T. Hirose, K. Tsushima and K. Umeda (Sumitomo Chemical Company, Limited): PCT Int. Appl. WO 96/11909 (1996).
- 53) N. Sakamoto, S. Saito, T. Hirose, M. Suzuki, S. Matsuo, K. Izumi, T. Nagatomi, H. Ikegami, K. Umeda, K. Tsushima and N. Matsuo: The discovery of pyridalyl: A novel insecticidal agent for controlling lepidopterous pests. Pest Manag. Sci. 60, 25–34 (2003).
- 54) S. Saito, S. Isayama, N. Sakamoto and K. Umeda: Insecticidal activity of pyridalyl: Acute and sub-acute symptoms in Spodoptera litura Larvae. J. Pestic. Sci. 29, 372–375 (2004).
- 55) H. Sakaguchi and M. Sasaki: (Sumitomo Chemical Company, Limited): US Pat. Appl. US 6590104 (2003).
- 56) M. R. Loso, B. M. Nugent, J. X. Huang, R. B. Rogers, Y. Zhu, J. M. Renga, V. B. Hegde, J. J. DeMark (Dow AgroSciences LLC): PCT Int. Appl. WO 2007/095229 (2007).
- 57) Y. Zhu, M. R. Loso, B. M. Nugent, J. X. Huang and R. B. Rogers: (Dow AgroSciences LLC): US Pat. Appl. US 20080108667 (2008).
- 58) Y. Zhu, M. R. Loso, G. B. Watson, T. C. Sparks, R. B. Rogers, J. X. Huang, B. C. Gerwick, J. M. Babcock, D. Kelly, V. B. Hegde, B. M. Nugent, J. M. Renga, I. Denholm, K. Gorman, G. J. DeBoer, J. Hasler, T. Meade and J. D. Thomas: Discovery and Characterization of Sulfoxaflor, a Novel Insecticide Targeting Sap-Feeding Pests. J. Agric. Food Chem. 59, 2950–2957 (2011).
- 59) T. C. Sparks, G. B. Watson, M. L. Loso, C. Geng, J. M. Babcock and J. D. Thomas: Sulfoxaflor and the sulfoximine insecticides: Chemistry, mode of action and basis for efficacy on resistant insects. Pestic. Biochem. Physiol. 107, 1–7 (2013).
- 60) K. E. Arndt, D. C. Bland, N. M. Irvine, S. L. Powers, T. P. Martin, J. R. McConnell, D. E. Podhorez, J. M. Renga, R. Ross, G. A. Roth, B. D. Scherzer and T. W. Toyzan: Development of a Scalable Process for the Crop Protection Agent Isoclast. Org. Process Res. Dev. 19, 454–462 (2015).
- 61) G. P. Lahm, R. M. Lett, B. T. Smith, B. K. Smith, C. A. Tyler (E. I. du Pont de Nemours and Company): PCT Int. Appl. WO 2010/129500 (2010).
- 62) G. P. Lahm, J. Desaeger, B. K. Smith, T. F. Pahutski, M. A. Rivera, T. Meloro, R. Kucharczyk, R. M. Lett, A. Daly, B. T. Smith, D. Cordova, T. Thoden and J. A. Wiles: The discovery of fluazaindolizine: A new product for the control of plant parasitic nematodes. Bioorg. Med. Chem. Lett. 27, 1572–1575 (2017).
- 63) I. Hamamoto, K. Koizumi, M. Kawaguchi, H. Tanigawa, T. Nakamura and T. Kobayashi (Nippon Soda Co., Ltd.): PCT Int. Appl. WO 2011/105506 (2011).
- 64) C. J. Urch, T. Lewis, R. L. Sunley, R. Salmon, C. R. Godfrey, C. I. Brightwell and M. B. Hotson (Zeneca Limited): PCT Int. Appl. WO 98/25924 (1998).
- 65) M. Kawaguchi: Abstr. Symp. 36th Res Committee for the Bioactivity of Pesticides, Pestic. Sci. Soc. Jpn, 21–24 (2019) (in Japanese).
- 66) A. C. O’sullivan, O. Loiseleur, R. Staiger, T. Luksch and T. Pitterna (Syngenta Participations AG): PCT Int. Appl. WO 2013/143811 (2013).
- 67) A. C. O’sullivan, R. J. G. Mondiere, O. Loiseleur, T. Smejkal, T. Luksch, A. Jeanguenat, R. Dumeunier, E. Godineau and T. Pitterna (Syngenta Participations AG): PCT Int. Appl. WO 2015/003951 (2015).
- 68) R. Dumeunier, T. Smejkal, B. P. Mishra, V. R. Gopalsamuthiram, E. Godineau and A. C. O’sullivan (Syngenta Participations AG): PCT Int. Appl. WO 2019/096860 (2019).
- 69) F. Erver and D. Brohm (Bayer Aktiengesellschaft): PCT Int. Appl. WO 2019/224174 (2019).
- 70) F. Cottet, M. Marull, O. Lefebvre and M. Schlosser: Recommendable Routes to Trifluoromethyl-Substituted Pyridine- and Quinolinecarboxylic Acids. Eur. J. Org. Chem. 2003, 1559–1568 (2003).
- 71) A. Lishchynskyi, M. A. Novikov, E. Martin, E. C. Escudero-Adán, P. Novák and V. V. Grushin: Trifluoromethylation of Aryl and Heteroaryl Halides with Fluoroform-Derived CuCF3: Scope, Limitations, and Mechanistic Features. J. Org. Chem. 78, 11126–11146 (2013).
- 72) L. E. Kiss, H. S. Ferreira and D. A. Learmonth: Efficient synthesis of 2-(Trifluoromethyl)nicotinic Acid Derivatives from Simple Fluorinated Precursors. Org. Lett. 10, 1835–1837 (2008).
- 73) R. Nishiyama, K. Fujikawa, T. Haga, T. Toki, K. Nagatani and O. Imai (Ishihara Sangyo Kaisha, Ltd.): Eur. Pat. Appl. EP 0031257 (1981).
- 74) FRAC: https://www.frac.info/docs/default-source/publications/frac-code-list/frac-code-list-2021--final.pdf?sfvrsn=f7ec499a_2 (Accessed Apr. 28, 2021).
- 75) T. Akagi, S. Mitani, T. Komyoji and K. Nagatani: Quantitative Structure–Activity Relationships of Fluazinam and Related Fungicidal N-Phenylpyridinamines: Preventive Activity against Botrytis cinerea. J. Pestic. Sci. 20, 279–290 (1995).
- 76) J. M. Clough, C. R. A. Godfrey, P. J. de Fraine, M. G. Hutchings and V. M. Anthony (Imperial Chemical Industries): Eur. Pat. Appl. EP 0278595 (1988).
- 77) F. E. Torba and C. Calif: (The Dow Chemical Company): US Pat. Appl. US 3609158 (1971).
- 78) P. J. de Fraine, M. C. Bowden and P. McNeilly: (Zeneca Limited): UK Pat. Appl. GB 2305174 (1997).
- 79) S. M. Brown, M. C. Bowden, T. J. Parsons, P. McNeilly and P. J. de Fraine: 6-(Trifluoromethyl)pyrid-2-one: Development and Scale-Up of a Ring Synthesis Route Based on Trifluoroacetic Anhydride. Org. Process Res. Dev. 1, 370–378 (1997).
- 80) G. Briggs, D. Mansfield, B. Moloney, S. Gary and T. Wegmann: The discovery and chemistry of fluopicolide: A new standard for oomycetes disease control. Pflanzenschutz-Nachr. Bayer 59, 141–152 (2006).
- 81) P. Jeschke: Progress of modern agricultural chemistry and future prospects. Pest Manag. Sci. 72, 433–455 (2016).
- 82) T. R. Faske and K. Hurd: Sensitivity of Meloidogyne incognita and Rotylenchulus reniformis to Fluopyram. J. Nematol. 47, 316–321 (2015).
- 83) B. A. Moloney, D. Hardy, E. A. Saville-Stones (AgrEvo UK Ltd.): PCT Int. Appl. WO 99/42447 (1999).
- 84) D. J. Mansfield, T. Cooke, P. S. Thomas, P. Y, Coqueron, J. P. Vors, G. G. Briggs, H. Lachaise, H. Rieck, P. Desbordes and M. C. Grosjean-Cournoyer (Bayer Cropscience S.A.): PCT Int. Appl. WO2004/016088 (2004).
- 85) J. Tang and A. Wang (Shandong United Pesticide Industry Co., Ltd.): Chinese Pat. Appl. CN 102086173 (2011).
- 86) R. Zhang, H. Y. Wang, H. Xv, J. Wang and K. Y. Wang: Uptake and transportation behavior of a new fungicidal agent LH-2010A in cucumber plants. J. Pestic. Sci. 39, 43–47 (2014).
- 87) X. Ji, J. Li, Z. Meng, N. Li, B. Dong, S. Zhang and K. Qiao: Fluopimomide effectively controls Meloidogyne incognita and shows a growth promotion effect in cucumber. J. Pestic. Sci. 93, 1421–1430 (2020).
- 88) D. O’Hagan: Fluorine in health care: Organofluorine containing blockbuster drugs. J. Fluor. Chem. 131, 1071–1081 (2010).
- 89) M. Inoue, Y. Sumii and N. Shibata: Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 5, 10633–10640 (2020).
- 90) https://adisinsight.springer.com/ (Accessed Feb. 8, 2021)
- 91) S. R. Turner, J. W. Strohbach, R. A. Tommasi, P. A. Aristoff, P. D. Johnson, H. I. Skulnick, L. A. Dolak, E. P. Seest, P. K. Tomich, M. J. Bohanon, M. M. Horng, J. C. Lynn, K. T. Chong, R. R. Hinshaw, K. D. Watenpaugh, M. N. Janakiraman and S. Thaisrivongs: Tipranavir (PNU-140690): A Potent, Orally Bioavailable Nonpeptidic HIV Protease Inhibitor of the 5,6-Dihydro-4-hydroxy-2-pyrone Sulfonamide Class. J. Med. Chem. 41, 3467–3476 (1998).
- 92) K. R. Romines, G. L. Bundy, R. A. Tommasi, J. W. Strohbach, S. R. Turner, S. Thaisrivongs, P. A. Aristoff, P. D. Johonson, H. I. Skulnick, L. L. Skaletzki, D. J. Anderson, J. Morris, R. B. Gammill and G. P. Luke (The Upjohn Company): PCT Int. Appl. WO 95/30670 (1995).
- 93) T. M. Judge, G. Phillips, J. K. Morris, K. D. Lovasz, K. R. Romines, G. P. Luke, J. Tulinsky, J. M. Tustin, R. A. Chrusciel, L. A. Dolak, S. A. Mizsak, W. Watt, J. Morris, S. L. Vander Velde, J. W. Strohbach and R. B. Gammill: Asymmetric Syntheses and Absolute Stereochemistry of 5,6-Dihydro-α-pyrones, A New class of potent HIV Protease Inhibitors. J. Am. Chem. Soc. 119, 3627–3628 (1997).
- 94) B. M. Trost and N. G. Andersen: Utilization of Molybdenum- and Palladium-Catalyzed Dynamic Kinetic Asymmetric Transformations for the prparation of Tertiary and Quaternary Stereogenic Centers: A Concise Synthesis of Tipranavir. J. Am. Chem. Soc. 124, 14320–14321 (2002).
- 95) G. Cianchetta, B. Delabarre, J. Popovicimuller, F. G. Salituro, J. O. Saunders, J. Travins, S. Yan, T Guo and L. Zhang (Agios Pharmaceuticals, Inc.): PCT Int. Appl. WO 2013/102431 (2013).
- 96) R. Dogra, R. Bhatia, R. Shankar, P. Bansal and R. K. Rawal: Enasidenib: First Mutant IDH2 Inhibitor for the Treatment of Refractory and Relapsed Acute Myeloid Leukemia. Anti-Cancer. Agents Med. Chem. 18, 1936–1951 (2018).
- 97) M. M. Kremlev, A. I. Mushta, W. Tyrra, Y. L. Yagupolskii, D. Naumann and A. Möller: Me3SiCF3/AgF/Cu—A new reagents combination for selective trifluoromethylation of various organic halides by trifluoromethylcopper, CuCF3. J. Fluor. Chem. 133, 67–71 (2012).
- 98) S. E. Webber, T. M. Bleckman, J. Attard, J. G. Deal, V. Kathardekar, K. M. Welsh, S. Webber, C. A. Janson, D. A. Matthews, W. W. Smith, S. T. Freer, S. R. Jordan, R. J. Bacquet, E. F. Howland, C. L. J. Booth, R. W. Ward, S. M. Hermann, J. White, C. A. Morse, J. A. Hilliard and C. A. Bartlett: Design of Thymidylate Synthase Inhibitors Using Protein Crystal Structures: The Synthesis and Biological Evaluation of a Novel Class of 5-Substituted Quinazolinones. J. Med. Chem. 36, 733–746 (1993).
- 99) M. E. Jung, C. L. Sawyers, S. Ouk, C. Tran and J. Wongvipat (The Regents of The University of California): PCT Int. Appl. WO2007/126765 (2007).
- 100) P. Rajaram, A. Rivera, K. Muthima, N. Olveda, H. Muchalski and Q. H. Chen: Second-Generation Androgen Receptor Antagonists as Hormonal Therapeutics for Three Forms of Prostate Cancer. Molecules 25, 2448 (2020).
- 101) D. L. Hughes: Review of Synthetic Routes and Crystalline Forms of the Antiandrogen Oncology Drugs Enzalutamide, Apalutamide, and Darolutamide. Org. Process Res. Dev. 24, 347–362 (2020).
- 102) J. Burch, B. Cote, N. Nguyen, C. S. Li, M. St-Onge and D. Gauvreau (Merck Frosst Canada Ltd.): PCT Pat. Appl. WO2011/120133 (2011).
- 103) R. Talwani and Z. Temesgen: Doravirine: a new non-nucleoside reverse transcriptase inhibitor for the treatment of HIV infection. Drugs Today (Barc) 56, 113–124 (2020).
- 104) B. Côté, J. D. Burch, E. Asante-Appiah, C. Bayly, L. Bédard, M. Blouin, L. C. Campeau, E. Cauchon, M. Chan, A. Chefson, N. Coulombe, W. Cromlish, S. Debath, D. Deschênes, K. Dupont-Gaudet, J. P. Falgueyret, R. Forget, S. Gagné, D. Gauvreau, M. Girardin, S. Guiral, E. Langlois, C. S. Li, N. Nguyen, R. Papp, S. Plamondon, A. Roy, S. Roy, R. Seliniotakis, M. St-Onge, S. Ouellet, P. Tawa, J. F. Truchon, J. Vacca, M. Wrona, Y. Yan and Y. Ducharme: Discovery of MK-1439, an orally bioavailable non-nucleoside reverse transcriptase inhibitor potent against a wide range of resistant mutant HIV viruses. Bioorg. Med. Chem. Lett. 24, 917–922 (2014).
- 105) V. Namasivayam, M. Vanangamudi, V. G. Kramer, S. Kurup, P. Zhan, X. Liu, J. Kongsted and S. N. Byrareddy: The Journey of HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs) from Lab to Clinic. J. Med. Chem. 62, 4851–4883 (2019).
- 106) L. C. Campeau, Q. Chen, D. Gauvreau, M. Girardin, K. Belyk, P. Maligres, G. Zhou, C. Gu, W. Zhang, L. Tan and P. D. O’shea: A Robust Kilo-Scale Synthesis of Doravirine. Org. Process Res. Dev. 20, 1476–1481 (2016).
- 107) D. R. Gauthier Jr., B. D. Sherry, Y. Cao, M. Journet, G. Humphrey, T. Itoh, I. Mangion and D. M. Tschaen: Highly Efficient Synthesis of HIV NNRTI Doravirine. Org. Lett. 17, 1353–1356 (2015).
- 108) C, Zhang, J. Zhang, P. N. Ibrahim, D. R. Artis, R. Bremer, G. Wu, H. Zhu and M. Nespi (Plexxikon, Inc.): PCT Int. Appl. WO 2008/064265 (2008).
- 109) M. I. El-Gamal, S. K. Al-Ameen, D. M. Al-Koumi, M. G. Hamad, N. A. Jalal and C. H. Oh: Recent Advances of Colony-Stimulating Factor-1 Receptor (CSF-1R) Kinase and Its Inhibitors. J. Med. Chem. 61, 5450–5466 (2018).
- 110) D. Chen, Y. Zhang, J. Li and Y. Liu: Exploratory Process Development of Pexidartinib through the Tandem Tsuji–Trost Reaction and Heck Coupling. Synthesis 51, 2564–2571 (2019).
- 111) P. Mueller (Bayer AG): Eur. Pat. Appl. EP 1340747 (2003).
- 112) E. K. Bayburt, B. Clapham, P. B. Cox, J. F. Daanen, M. J. Dart, G. A. Gfesser, A. Gomtsyan, M. E. Kort, P. R. Kym, E. A. Voight and R. G. Schmidt (AbbVie Inc.): PCT Int. Appl. WO2013/062966 (2013).
- 113) G. Bernasconi, S. M. Bromidge, A. J. Carpenter, L. D’adamo, R. Di Fabio, S. Guery, F. Pavone, A. Pozzan, M. Rinaldi, F. M. Sabbatini and Y. St-Denis (Glaxo Group Limited): PCT Int. Appl. WO2008/148853 (2008).
- 114) M. A. Brondney, J. E. Davoren, A. B. Dounay, I. V. Efremov, D. L. F. Gray, M. E. Green, J. L. Henderson, C. Lee, S. R. Mente, S. V. O’Neil, B. N. Roger and L. Zhang (Pfizer Inc.): PCT Int. Appl. WO 2014/207601 (2014).
- 115) D. Young, M. Popiolek, P. Trapa, K. R. Fonseca, J. Brevard, D. L. Gray and R. Kozak: D1 Agonist Improved Movement of Parkinsonian Nonhuman Primates with Limited Dyskinesia Side Effects. ACS Chem. Neurosci. 11, 560–566 (2020).
- 116) N. G. Cooke, P. Fernandes Gomes Dos Santos, N. Graveleau, C. Hebach, K. Högenauer, G. Hollingworth, A. B. Smith, N. Soldermann, F. Stowasser, R. Strang, N. Tufilli, A. Von Matt, R. Wolf and F. Zecri (Novaltis AG): PCT Int. Appl. WO2012/004299 (2012).
- 117) K. Hoegenauer, N. Soldermann, F. Zécri, R. S. Strang, N. Graveleau, R. M. Wolf, N. G. Cooke, A. B. Smith, G. J. Hollingworth, J. Blanz, S. Gutmann, G. Rummel, A. Littlewood-Evans and C. Burkhart: Discovery of CDZ173 (Leniolisib), Representing a Structurally Novel Class of PI3K Delta-Selective Inhibitors. ACS Med. Chem. Lett. 8, 975–980 (2017).
- 118) H. Hunt, T. Johnson, N. Ray and I. Walters (Corcept Therapeutics, Inc.): PCT Int. Appl. WO2013/177559 (2013).
- 119) H. J. Hunt, J. K. Belanoff, I. Walters, B. Gourdet, J. Thomas, N. Barton, J. Unitt, T. Phillips, D. Swift and E. Eaton: Identification of the Clinical Candidate (R)-(1-(4-Fluorophenyl)-6-((1-methyl-1H-pyrazol-4-yl)sulfonyl)-4,4a,5,6,7,8-hexahydro-1H-pyrazolo[3,4-g]isoquinolin-4a-yl)(4-(trifluoromethyl)pyridin-2-yl)methanone (CORT125134): A Selective Glucocorticoid Receptor (GR) Antagonist. J. Med. Chem. 60, 3405–3421 (2017).
- 120) B. Lehnemann, J. Jung and A. Meudt (Archimica GmbH): PCT Int. Appl. WO 2007/000249 (2007).
- 121) V. Cmiljanovic, N. Cmiljanovic, B. Giese and M. Wymann (University of Basel): PCT Int. Appl. WO2010/052569 (2010).
- 122) F. Beaufils, N. Cmiljanovic, V. Cmiljanovic, T. Bohnacker, A. Melone, R. Marone, E. Jackson, X. Zhang, A. Sele, C. Borsari, J. Mestan, P. Hebeisen, P. Hillmann, B. Giese, M. Zvelebil, D. Fabbro, R. L. Williams, D. Rageot and M. P. Wymann: 5-(4,6-Dimorpholino-1,3,5-triazin-2-yl)-4-(trifluoromethyl)pyridin-2-amine (PQR309), a Potent, Brain-Penetrant, Orally Bioavailable, Pan-Class I PI3K/mTOR Inhibitor as Clinical Candidate in Oncology. J. Med. Chem. 60, 7524–7538 (2017).
- 123) P. Hebeisen, F. Beaufils and J-B. Langlois (Universitaet Basel, Switz., PIQUR Therapeutics AG): PCT Int. Appl. WO2015/162084 (2015).
- 124) U. Baetting, J. K. Bala, E. Budd, L. Edwards, C. Howsham, G. Hughes, M. D. Legrsnd and K. Spiegel (Novartis AG): PCT Int. Appl. WO2011/113894 (2011).
- 125) T. Heinz, B. Martin, A. F. Rampe and W. Zaugg (Novartis AG): PCT Int. Appl. WO2018/116139 (2018).
- 126) T. Kawabe, M. Ishigaki, T. Sato, S. Yamamoto and Y. Hasegawa (CanBas Co.,Ltd.): US. Pat. Appl. US 2008/0275057 (2008).
- 127) K. Sakakibara, N. Saito, T. Sato, A. Suzuki, Y. Hasegawa, J. M. Friedman, D. W. Kufe, D. D. VonHoff, T. Iwami and T. Kawabe: CBS9106 is a novel reversible oral CRM1 inhibitor with CRM1 degrading activity. Blood 118, 3922–3931 (2011).
- 128) G. A. Nishiguchi, A. Rico, H. Tanner, R. J. Aversa, B. R. Taft, S. Subramanian, L. Setti, M. T. Burger, L. Wan, V. Tamez, A. Smith, Y. Lou, P. A. Barsanti, B. A. Appleton, M. Mamo, L. Tandeske, I. Dix, J. E. Tellew, S. Huang, L. A. Mathews Griner, V. G. Cooke, A. Van Abbema, H. Merritt, S. Ma, K. Gampa, F. Feng, J. Yuan, Y. Wang, J. R. Haling, S. Vaziri, M. Hekmat-Nejad, J. M. Jansen, V. Polyakov, R. Zang, V. Sethuraman, P. Amiri, M. Singh, E. Lees, W. Shao, D. D. Stuart, M. P. Dillon and S. Ramurthy: Design and Discovery of N-(2-Methyl-5′-morpholino-6′-((tetrahydro-2H-pyran-4-yl)oxy)-[3,3′-bipyridin]-5-yl)-3-(trifluoromethyl)benzamide (RAF709): A Potent, Selective, and Efficacious RAF Inhibitor Targeting RAS Mutant Cancers. J. Med. Chem. 60, 4869–4881 (2017).
- 129) R. Aversa, P. A. Barsanti, M. Burger, M. P. Dillon, A. Dipesa, C. Hu, Y. Lou, G. Nishiguchi, Y. Pan, V. Polyakov, S. Ramurthy, A. Rico, L. Setti, A. Smith, S. Subramanian, B. Taft, H. Tanner, L. Wan and N. Yusuff (Novaltis AG): PCT Pat. Appl. WO2014/151616 (2014).
- 130) S. Ramurthy, B. R. Taft, R. J. Aversa, P. A. Barsanti, M. T. Burger, Y. Lou, G. A. Nishiguchi, A. Rico, L. Setti, A. Smith, S. Subramanian, V. Tamez, H. Tanner, L. Wan, C. Hu, B. A. Appleton, M. Mamo, L. Tandeske, J. E. Tellew, S. Huang, Q. Yue, A. Chaudhary, H. Tian, R. Lyer, A. Q. Hassan, L. A. M. Griner, L. R. La Bonte, V. G. Cooke, A. V. Abbema, H. Merritt, K. Gampa, F. Feng, J. Yuan, Y. Mishina, Y. Wang, J. R. Haling, S. Vaziri, M. Hekmat-Nejad, V. Polyakov, R. Zang, V. Sethuraman, P. Amiri, M. Singh, W. R. Sellers, E. Lees, W. Shao, M. P. Dillon and D. D. Stuart: Design and Discovery of N-(3-(2-(2-Hydroxyethoxy)-6-morpholinopyridin-4-yl)-4-methylphenyl)-2-(trifluoromethyl)isonicotinamide, a Selective, Efficacious, and Well-Tolerated RAF Inhibitor Targeting RAS Mutant Cancers: The path to the Clinic. J. Med. Chem. 63, 2013–2027 (2020).
- 131) V. Maywald, F. Menges, J. U. Vogelbacher, M. Rack, M. Keil, W. Grammenos, M. Vrettou-Schultes, K. J. Lohmann, B. Mueller, T. Jabs (BASF SE) PCT Int. Appl. WO 2011/161612 (2011).
- 132) R. Trussardi (F. Hoffmann-La Roche AG) PCT Int. Appl. WO2014/076127 (2014).
- 133) M. Böger and G. S, Wilhelm (Ciba-Geigy AG): Eur. Pat. Appl. EP 0079311 (1983).
- 134) P. Junquera, B. Hosking, M. Gameiro and A. Macdonald: Benzoylphenyl ureas as veterinary antiparasitics. An overview and outlook with emphasis on efficacy, usage and resistance. Parasite 26, 26 (2019).
- 135) S. Yotsuya, H. Shikama, I. Nakano, K. Sakai, M. Kato, H. Sugi, H. Takada and Y. Koga: A Novel Synthetic Anti-Acute Pancreatitis Agents, IS-741. Digestion 60(Suppl 1), 34–39 (1999).
- 136) https://www.iskweb.co.jp/eng/products/animal_health.html (Accessed Nov. 10, 2020)
- 137) H. Kimura, S. Yotsuya, S. Yuki, H. Sugi, I. Shigehara and T. Haga: Synthesis and Antipancreatitis Activities of Novel N-(2-Sulfonylamino-5-trifluoromethyl-3-pyridyl) carboxamide Derivatives as Phospholipase A2 Inhibitors. Chem. Pharm. Bull. 43, 1696–1700 (1995).
- 138) D. O’Hagan and D. B. Harper: Fluorine-containing natural products. J. Fluor. Chem. 100, 127–133 (1999).
- 139) R. M. Asath, R. Premkumar, T. Mathavan and A. M. F. Benial: Structural, spectroscopic and molecular docking studies on 2-amino-3-chloro-5-trifluoromethyl pyridine: A potential bioactive agent. Spectrochim. Acta A Mol. Biomol. Spectrosc. 175, 51–60 (2017).
- 140) P. Robatscher, D. Eisenstecken, G. Innerebner, C. Roschatt, B. Raifer, H. Rohregger, H. Hafner and M. Oberhuber: 3-Chloro-5-trifluoromethylpyridine-2-carboxylic acid, a Metabolite of the Fungicide Fluopyram, Cause Growth Disorder in Vitis vinifera. J. Agric. Food Chem. 67, 7223–7231 (2019).