Abstract
Since chloroplasts and mitochondria are maternally inherited and have unique features in evolution, DNA sequences of those organelle genomes have been broadly used in phylogenetic studies. Thanks to recent progress in next-generation sequencer (NGS) technology, whole-genome sequencing can be easily performed. Here, using NGS data generated by Roche GS Titanium and Illumina Hiseq 2000, we performed a hybrid assembly of organelle genome sequences of Vigna angularis (azuki bean). Both the mitochondrial genome (mtDNA) and the chloroplast genome (cpDNA) of V. angularis have very similar size and gene content to those of V. radiata (mungbean). However, in structure, mtDNA sequences have undergone many recombination events after divergence from the common ancestor of V. angularis and V. radiata, whereas cpDNAs are almost identical between the two. The stability of cpDNAs and the variability of mtDNAs was further confirmed by comparative analysis of Vigna organelles with model plants Lotus japonicus and Arabidopsis thaliana.
Introduction
Genus Vigna contains several cultivated species such as cowpea and mungbean that are important protein sources in Africa and Asia. In addition, the wild species of genus Vigna are potentially important genetic resources because of their outstanding adaptability to severe environmental conditions such as saline, arid, acidic or alkaline soils (Maxted et al. 2004, Tomooka et al. 2002). Thus, to understand the evolutionary dynamics and adaptation mechanisms of Vigna species, a genome project that aims at sequencing whole genomes of 16 species including both cultivated and wild species has been initiated. This project includes sequencing the chloroplast and mitochondrial genomes (cpDNAs and mtDNAs, respectively), because organelles are maternally inherited, have unique features in evolution and thus are important for reconstructing the phylogenetic relationships among organisms (Jansen et al. 2005).
This genome project has started with azuki bean [V. angularis (Willd.) Ohwi & Ohashi], because it is an important traditional grain legume in East Asia. In addition, it has considerable cultural importance in Japan, Korea and northern and central China (Lumpkin and McClary 1994) and the traditional cultivation area extends through southern China to the Himalayan foothills of Bhutan, India and Nepal (Tomooka et al. 2002).
Here in this study, we report a de novo assembly of the organelle genomes of azuki bean using the next-generation sequencer (NGS) data. Sequencing of mtDNAs has been a challenge because of its frequent intra- and inter-molecular recombination, however, deep coverage achieved by NGS data enabled to distinguish the original sequences from recombination products. We compared azuki bean organelle genomes with those of V. radiata (mungbean) (Alverson et al. 2011, Tangphatsornruang et al. 2010) to elucidate evolutionary dynamics of cpDNAs and mtDNAs within Vigna. Furthermore, because whole genome alignment of mtDNAs among plant species has not often been performed, we compared the Vigna mtDNA with those of Lotus japonicus and Arabidopsis thaliana to determine how quickly mtDNA sequences evolve.
Materials and Methods
Plant materials and DNA extraction
Seeds of V. angularis cv. ‘Shumari’ were provided by Tokachi Agricultural Experiment Station of Hokkaido Research Organization, Memuro, Hokkaido, Japan. For the first run of Roche GS FLX Titanium, total DNA was extracted from the leaves of 1 week old plants using DNeasy Plant Mini Kit (Qiagen K. K., Tokyo). For the second run and subsequent runs, we extracted nuclei-condensed DNA using the percoll method described by Henfrey and Slater (1988), since too much organelle DNA was found in the data of the first run. The nuclei-condensed DNA still contains some organelle DNA and could be used for organelle genome assembly.
DNA sequencing
For the Roche GS FLX platform, construction of mate-pair libraries and sequencing were all provided as a custom service of Beckman Coulter Genomics (Danvers, MA, USA). In total, we performed 4 runs for 3 kb mate-pair library, 4 runs for 8 kb mate-pair library and 3 runs for 20 kb mate-pair library.
For the Illumina Hiseq 2000 platform, library construction and sequencing was provided as a custom service of Eurofins MWG GmbH, Ebersberg, Germany. Sequencing libraries included paired-end library of 300 bp insert and mate-pair libraries with insert sizes of 8 kb, 20 kb and 40 kb. One lane of the flow cell was used for each sequencing library.
De novo assembly of NGS data
The obtained sequence reads of Roche GS FLX Titanium were assembled using the de novo assembly program of genomic workbench software (CLC bio, Aarhus, Denmark). We then sorted the assembled contiguous sequences (contigs) by depth of the coverage to classify those into cpDNA, mtDNA and nuclear genomes: 2,000–3,000× for cpDNA, ~200× for mtDNA and ~20× for nucleus. We confirmed those with high-coverage (more than 100x) derive from organelle DNAs by BLASTing against mungbean cpDNA and mtDNA sequences and then proceeded to the following process.
Most de novo assembly programs, including the one in genomic workbench software, are graph-based program, which finds overlaps of the reads and merges into contigs. However, at the boundaries of repeat sequences, for example, multiple ways of extending contigs would be detected. Thus, the assembly programs cut off the contigs at such “edged” sites (reviewed by Henson et al. 2012). Because mtDNA undergoes frequent recombination at various sites, the assemblies of mtDNA often encounter such conflicting edges of the contigs. To solve this problem, we collected reads overlapping the edges and then assembled using a classical “greedy” program. This process typically produces four contigs: two with original sequences (higher coverage) and two with recombinant products (very low coverage). We took the high-coverage contigs as sequences of the master-circle and then reassembled into super-contigs. The coverage data of the original sequences and the recombinant products were also used to estimate the numerical rate of recombination of mtDNAs in azuki bean cells. Non-recombining repeat sequences were also detected by BLASTing the mtDNA sequence to itself.
Read mapping and correction of draft genome sequences
After the circular draft genomes of the mtDNA and the cpDNA were reconstructed, we mapped the Illumina reads as well as GS FLX reads to the draft genomes by CLC genomic workbench software to find sites of misassemblies and to correct the errors including homopolymers. Mis-assemblies can be detected as gaps or mate-paired reads with wrong directions or unexpected insert sizes. Therefore the draft genomes at the misassembled sites were broken and then repeated the contig-extension process described above. Reassembled sequences were again checked by read-mapping. After all the errors and misassemblies were corrected, encoded genes in organelle genomes were manually annotated. The complete sequences of cpDNA (AP012598) and mtDNA (AP012599) are publicly available at DDBJ (DNA data bank of Japan, http://www.ddbj.nig.ac.jp/updt-form-j.html).
Comparative analysis of organelle genomes
The mtDNA and the cpDNA sequences of mungbean (V. radiata, GI:323149028 and GI:289066804, respectively), L. japonicus (GI:13518417 and GI:387866040, respectively) and A. thaliana (GI:7525012 and GI:26556996, respectively) were downloaded from NCBI nucleotide database. Dot-plot analysis was performed using Genome-Matcher with the default settings (Otsubo et al. 2008). Whole genome alignment and extraction of SNPs and small indels were performed using MUMmer3 software (Kurtz et al. 2004).
Results
cpDNA in azuki bean
Initially whole Roche GS FLX reads were used for de novo assembly of the azuki bean genome. However, the de novo assembly program of the CLC bio genomic workbench software did not seem to handle the mate-pair reads with 20 kb insert very well. Thus, the mate-pair reads of 3 kb and 8 kb inserts were used. Out of tens of thousands of contigs generated, cpDNA was assembled into only three contigs: large single copy (LSC), small single copy (SSC) and inverted-repeats (IRs). Since IRs contain only one SNP compared to each other (described later), they were merged into one contig. As expected, the coverage of the IR contig was twice as deep as the contigs of LSC and SSC. Thus, it was easy to reconstruct cpDNA: simply connect LSC, IR, SSC and reverse-complementary of IR (Fig. 1). The accuracy of the re-assembled super-contig was assessed by mapping the Illumina reads onto the super-contig and checking the mapped directions and the distances of the paired-reads. Homopolymers and other sequence errors in the super-contig were also corrected in this process. This process detected one SNP between the copies of IRs. PCR followed by Sanger-sequencing further confirmed the accuracy of the manually-closed gaps and the corrected sequences.
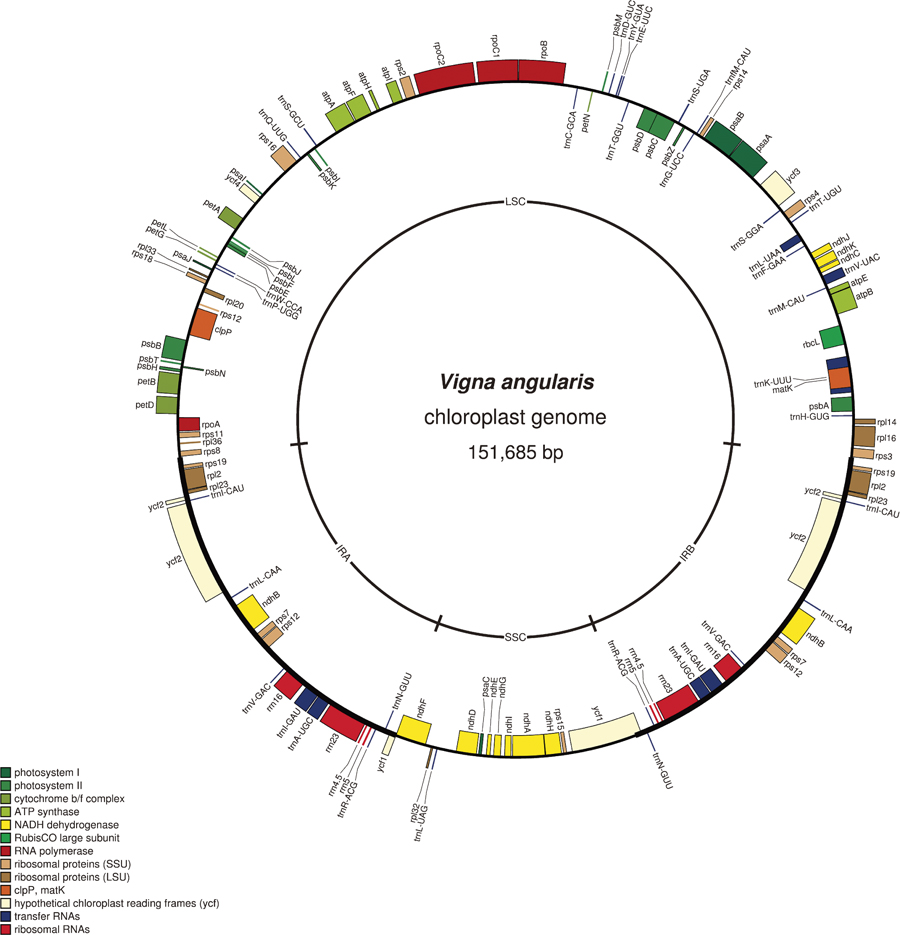
The constructed azuki cpDNA is 151,591 bp long in total, with 81,028 bp of LSC, 17,464 bp SSC plus a pair of IRs of 26,460 bp (Fig. 1). The cpDNA contained 108 unique genes, including 75 protein-coding genes, 4 rRNA genes and 29 tRNAs. Of these, 19 genes are duplicated in the IRs, making a total of 127 genes present in the cpDNA (Fig. 1, Supplemental Table 1). Although we did not have any transcriptional data, a presumed RNA-editing site in the ndhD gene was found to be conserved across the genus (Tangphatsornruang et al. 2010).
mtDNA in azuki bean
Compared to cpDNA, assembly of mtDNA was more complicated because mtDNA undergoes frequent recombination via even small repeat sequences. Thus, assembly programs cut off contigs at the sites of recombination even though read length is longer than the repeat sequences that serve as recombination substrate. For example, consider a mtDNA with two copies of a repeat sequence “R”. The single copy sequences flanking the one copy of “R” are “A” and “B” and those flanking the other are “C” and “D”, which makes up two concatenated sequences of “A-R-B” and “C-R-D”. After a recombination event, the two concatenated sequences turns into “A-R-D” and “C-R-B”. Because of these conflicting products, de novo assembly program cuts off the contigs at the boundaries of “R”. So, in this case, five contigs of “A”, “B”, “C”, “D” and “R” are obtained. The subsequent manual extension of the contig ends produces four super contigs of “A-R-B”, “A-R-D”, “C-R-B” and “C-R-D”. However, the original sequences and the recombinant products can be easily distinguished when Illumina reads are mapped onto these super-contigs. Although recombination is frequent in plant mtDNAs, the majority of mtDNA molecules in the cell still hold the original sequences. Thus, read-mapping results have a much higher coverage in the original sequences “A-R-B” and “C-R-D” than in the recombinant products “A-R-D” and “C-R-B”. The manually assembled draft sequence of the mtDNA was again read-mapped to correct any kinds of errors, as was performed for the cpDNA.
The reconstructed mtDNA is 404,446 bp long with 45% GC content, harboring 31 proteins, 3 ribosomal DNAs and 16 tRNA genes (Fig. 2, Supplemental Table 2). As for gene content, azuki bean mtDNA shares all the features observed in already-sequenced mungbean mtDNA (Alverson et al. 2011). Thus, compared to other plant mtDNAs, azuki bean mtDNA harbors two identical copies of atp9 but lacks cox2, rpl2, rpl10, rps2, rps11, rps13 and sdh3 genes and have rps7, rps19 and sdh4 as pseudo genes. Presumed RNA-editing sites in nad5, nad4L, rps10, nad1, mttB and ccmFc genes were also found to be conserved.
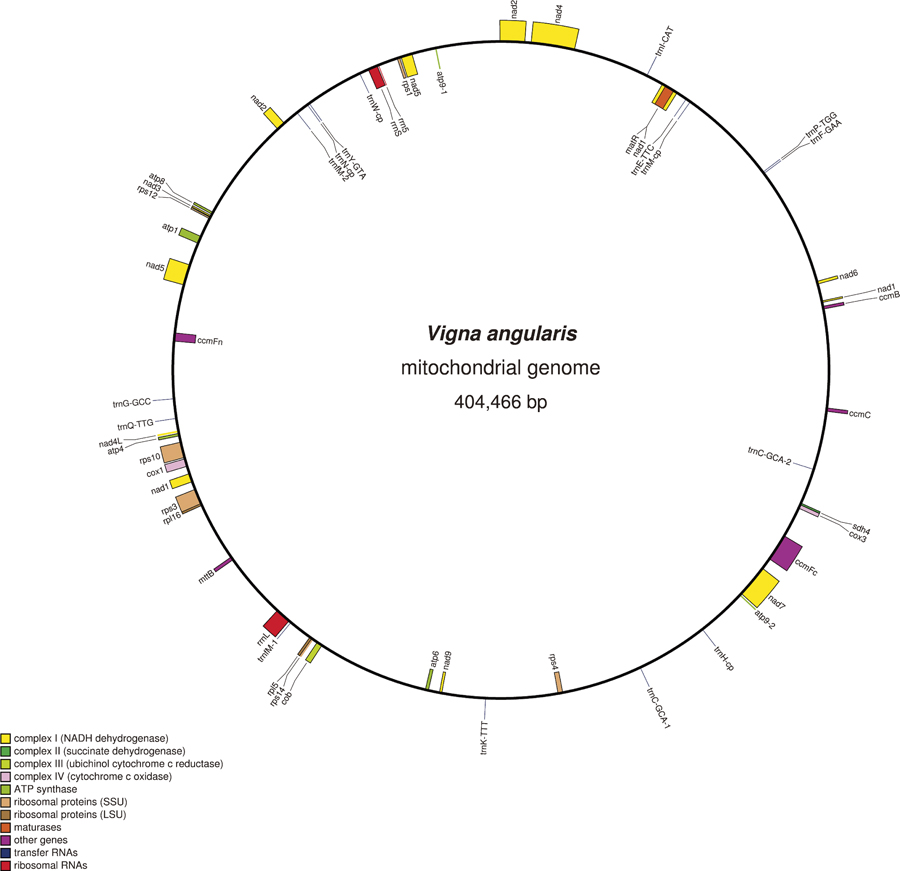
The depth of the read-mapping data of the original sequences and the recombinant products enabled us not only to detect recombination sites but also recombination frequency. All the recombination sites are repeat sequences ranging from 81 bp to 1,215 bp in length. Although there are many more repeat sequences (131 in total) in the mtDNA (Supplemental Table 3), no evidence of recombination was detected other than the twelve repeat elements shown in Table 1. Longer repeat sequences showed higher recombination rate, and direct repeats were more prone to recombination than inverted repeats.
Table 1
The recombinating repeat regions and recombination rates
| Repeat |
Length |
Copy |
Direction |
Position |
Recombination ratea |
| A |
1215 |
a |
+ |
404090..841 |
0.33 |
|
|
b |
+ |
193239..194455 |
|
| B |
110 |
a |
+ |
68512..68621 |
0.07 (a <=> b) |
|
|
b |
− |
210611..210720 |
|
|
|
c |
+ |
254685..254794 |
0.14 (c <=> d) |
|
|
d |
− |
361196..361305 |
|
| C |
62 |
a |
+ |
101337..101598 |
0.11 |
|
|
b |
− |
341474..341735 |
|
| D |
321 |
a |
+ |
113798..114118 |
0.08 |
|
|
b |
− |
355759..356079 |
|
| E |
137 |
a |
+ |
114018..114154 |
0.12 |
|
|
b |
− |
146537..146673 |
|
| H |
116 |
a |
+ |
114074..114189 |
0.09 |
|
|
b |
− |
402480..402595 |
|
| I |
204 |
a |
+ |
122790..122993 |
0.09 |
|
|
b |
− |
175077..175281 |
|
| J |
149 |
a |
+ |
177524..177672 |
0.09 |
|
|
b |
− |
289080..289228 |
|
| K |
235 |
a |
+ |
250286..250520 |
0.24 |
|
|
b |
+ |
341889..342123 |
|
| L |
123 |
a |
+ |
259471..259599 |
0.21 |
|
|
b |
+ |
276277..276396 |
|
| M |
103 |
a |
+ |
289083..289185 |
0.07 |
|
|
b |
+ |
344990..345101 |
|
| N |
81 |
a |
+ |
114074..114154 |
0.05 (a <=> c) |
|
|
b |
− |
146537..146617 |
|
|
|
c |
− |
402515..402595 |
|
a The recombining repeat copies are indicated in parentheses when there are three or more copies of the repeat sequences.
Because organelle genomes of another species of genus Vigna, V. radiata (mungbean), have been sequenced in previous studies (Alverson et al. 2011, Tangphatsornruang et al. 2010), we performed a comparative analysis of the organelle genomes between azuki bean and mungbean.
Although azuki bean cpDNA is 120 bp longer than mungbean, dot-plot analysis showed cpDNA structures are completely conserved between the two species, including IRs (Fig. 3A). The whole genome alignment by MUMmer3 program detected 1,034 SNPs, 92 insertions (present in azuki cpDNA but not in mungbean) and 108 deletions (absent in azuki bean) (Supplemental Table 4).

In contrast to cpDNAs, the dot-plot analysis of mtDNAs revealed evidence of frequent rearrangement that has divided Vigna’s mtDNA into 33 blocks (Fig. 3B). This result led us to doubt the quality of our assembly and thus we designed primers flanking the boundaries of the rearrangement to perform PCR. With azuki bean DNA as template PCR was positive for all primer pairs, while PCR was consistently negative with mungbean DNA. Thus, we concluded that the stochastic orders and orientations indicated by dot-plot analysis is not a result of misassembly. Although mtDNA structures are rearranged to a great extent, the identity within each synteny block is highly conserved even in intergenic regions. Scanning each homology block detected 2,215 SNPs, 117 insertions and 154 deletions (Supplemental Table 5).
Comparative analysis of mtDNAs between Vigna, Lotus and Arabidopsis
The previous study has reported a broad-ranged comparative analysis of cpDNAs of mungbean (V. radiata) and other plant species (Tangphatsornruang et al. 2010) and revealed a legume-specific inversion and a translocation specific to Phaseolinae (Phaseolus and Vigna) (see also Fig. 4). However, comparative analyses on mtDNAs have been limited because of its unstable features and lack of sequence data. For example, the complete mtDNA sequences are missing even in genome-sequenced species such as Glycine max (soybean) and Phaseolus vulgaris (common bean). Thus, before this study, mungbean and L. japonicus are the only legume species with mtDNAs sequenced.
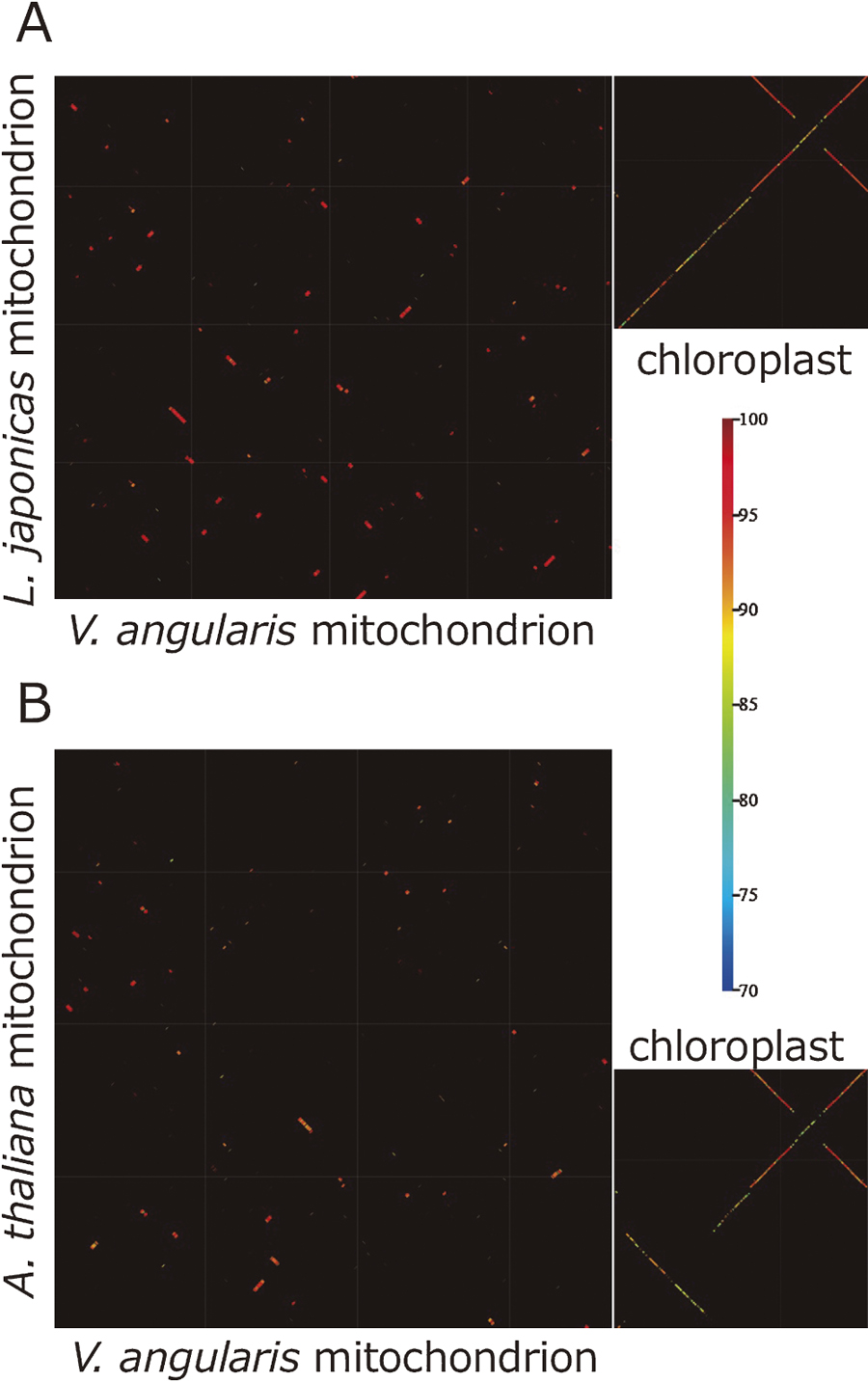
The drastic shuffling of mtDNAs between azuki bean and mungbean caused us to perform another whole mtDNA alignment between azuki bean and L. japonicus (Fig. 4A). As a result, only about 40% of mtDNA sequences could be aligned to each other. The aligned sequences were highly conserved and retained >95% identity, however, the remaining 60% had no homology at all. This contrasts with cpDNA dot-plot, where most of the sequences were well-aligned but identity of aligned sequences is 80–90% except IR regions (Fig. 4A). Furthermore, we aligned Arabidopsis thaliana’s mtDNA against azuki bean’s and found conserved sequences are limited only in CDS, while cpDNAs are still homologous to each other throughout the genome (Fig. 4B).
Discussion
In this study, we have determined the complete genome sequences of azuki bean organelles using only next-generation sequencers (NGS). Since there’s no perfect assembler program so far, de novo assembly processes always generate misassembled contigs. Thus, assembled contigs must be double-checked by read-mapping and be scanned for any gaps of lower coverage or of unexpected directions/distances of paired-reads. We also showed that this assemble-and-mapping process can resolve problems of frequent recombination in mtDNA.
Using the NGS data, we could also detect sites and frequency of recombination in the mtDNA of azuki bean (Table 1). The length of the repeats and recombination rate clearly correlated, and no trace of recombination was detected in repeat sequences of <100 bp except one. Recombination between direct repeats in a master-circle of mtDNA produces two mini-circles, while recombination between inverted repeats produces an inversion. The highest recombination rate (0.33) was observed between the longest-direct repeats (1,215 bp), indicating about one-third of the mtDNA molecules are divided into two mini-circles in azuki bean. With recombination between other repeats, mtDNAs in azuki bean may contain many inversions and even be subdivided into more mini-circles.
From the comparative analysis using organelle genomes, there are two points to be noted. One is the extreme stability of cpDNAs across plant taxa and the other is the rapid and massive changes in mtDNAs. The slow evolution of cpDNA is highlighted by the fact that there occurred only two structural changes during evolution of the Vigna lineage: One is the inversion between 8 kb–56 kb region which took place after Fabaceae ancestor has diverged from Brassicaceae, and the other is the translocation of 3 kb fragment after Vigna ancestors diverged from other legume species (Perry et al. 2002, Tangphatsornruang et al. 2010, See also Fig. 4). In contrast, mtDNAs greatly vary through rapid and stochastic rearrangement of organizations and replacement of intergenic sequences, though conserved sequences including CDS (and even RNA-editing sites) are identical.
These features indicate cpDNAs are much more suitable for phylogenetic studies for a broad range of diverse species, as suggested by Jansen et al. (2005). However, the quick change in structure of mtDNAs would greatly facilitate tracing the evolutionary dynamics of closely related species. Since our Vigna genome project will sequence 16 Vigna species in a few years, we are very interested in performing phylogenetic analysis on these valuable genetic resources using their mtDNA sequences.
Acknowledgements
This study was supported by Grants-in-Aid for Young Scientists (A) of Japan Society for the Promotion of Science.
Literature Cited
- Alverson, A. J., S. Zhuo, D.W. Rice, D.B. Sloan and J.D. Palmer (2011) The mitochondrial genome of the legume Vigna radiata and the analysis of recombination across short mitochondrial repeats. PLoS one 6(1): e16404.
- Henfrey, R.D. and R.J. Slater (1988) Isolation of plant nuclei. Methods Mol. Biol. 4: 447–542.
- Henson, J., G. Tischler and Z. Ning (2012) Next-generation sequencing and large genome assemblies. Pharmacogenomics 13: 901–915.
- Jansen, R.K., L.A. Raubeson, J.L. Boore, C.W. dePamphilis, T.W. Chumley, R.C. Haberle, S.K. Wyman, A.J. Alverson, R. Peery, S.J. Herman et al. (2005) Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol. 395:348–384.
- Kurtz, S., A. Phillippy, A.L. Delcher, M. Smoot, M. Shumway, C. Antonescu and S.L. Salzberg (2004) Versatile and open software for comparing large genomes. Genome Biol. 5: R12.
- Lumpkin, T.A. and D.C. McClary (1994) Azuki bean: botany, production and uses. CAB International, Wallingford UK.
- Maxted, N., P. Mabuza-Dlamini, H. Moss, S. Padulosi, A. Jarvis and L. Guarino (2004) An Ecogeographic Survey: African Vigna, International Plant Genetic Resources Institute, Rome, Italy.
- Ohtsubo, Y., W. Ikeda-Ohtsubo, Y. Nagata and M. Tsuda (2008) GenomeMatcher: a graphical user interface for DNA sequence comparison. BMC Bioinformatics 9: 376.
- Perry, A.S., S.B. Brennan, D.J. Murphy, T.A. Kavanagh and K.H. Wolfe (2002) Evolutionary re-organization of a large operon in adzuki bean chloroplast DNA caused by inverted repeat movement. DNA Res. 9: 157–162.
- Tangphatsornruang, S., D. Sangsrakru, J. Chanprasert, P. Uthaipaisanwong, T. Yoocha, N. Jomchai and S. Tragoonrung (2010) The chloroplast genome sequence of mungbean (Vigna radiata) determined by high-throughput pyrosequencing: structural organization and phylogenetic relationships. DNA Res. 17: 11–22.
- Tomooka, N., D.A. Vaughan, H. Mass and N. Maxted (2002) The Asian Vigna: genus Vigna subgenus Ceratotropis genetic resources, Kluwer Academic Publisher, Dordrecht, NL.