Abstract
Heterosis or hybrid vigor is a phenomenon where hybrid progeny have superior performance compared to their parental inbred lines. This is important in the use of F1 hybrid cultivars in many crops and vegetables. However, the molecular mechanism of heterosis is not clearly understood. Gene interactions between the two genomes such as dominance, overdominance, and epistasis have been suggested to explain the increased biomass and yield. Genetic analyses of F1 hybrids in maize, rice, and canola have defined a large number of quantitative trait loci, which may contribute to heterosis. Recent molecular analyses of transcriptomes together with reference to the epigenome of the parents and hybrids have begun to uncover new facts about the generation of heterosis. These include the identification of gene expression changes in hybrids, which may be important for heterosis, the role of epigenetic processes in heterosis, and the development of stable high yielding lines.
Introduction
In plants, F1 hybrids between specific combinations of parental lines show superior performance relative to the parental lines; the F1 hybrids have hybrid vigor or heterosis. The heterosis phenotype was first described in ‘The effects of cross and self-fertilization in the vegetable kingdom’ by Charles Darwin. George Harrison Shull introduced the term “heterosis” in 1914 to replace the more cumbersome word “heterozygosis”, which did not express the superior performance of the hybrids (Shull 1948). Heterosis is agronomically important because the superior performance can appear as biomass, yield, and abiotic and biotic stress tolerance. Breeding of F1 hybrid cultivars based on heterosis is used in many crops and vegetables.
In 1909, Shull found increased grain yields in hybrid corn and he proposed this as a method of corn breeding. His idea was accepted and led to the transition from open-pollinated to four-way cross hybrids (double cross hybrid), and then to two-way cross hybrids (single cross hybrid) (Crow 1998). In the Midwestern Corn Belt state of Iowa, hybrid corn was less than 10% of the crop in 1935, but it was over 90% four years later. Hybrid corn was the main crop in the 1950s in the United States, and yield advanced from one ton per hectare in 1930 to four tons per hectare in 1960 and approximately twelve tons per hectare in 2017. The reason for the rapid spreading of hybrid corn was due not only to increased yield, but also to uniformity in growth and flowering, which allowed machine harvesting (Crow 1998, Duvick 2001). Now, single cross hybrid corn has developed into a major industry worldwide. In the 1970s Yuan Longping developed hybrid rice with a significant yield advantage (10–20% greater) over inbred parental varieties. Hybrid rice was accepted in China, and about half of the total area of rice in China was planted to hybrids by the early 2000s (Cheng et al. 2007). Hybrids are also used in crops such as sorghum, sunflower, canola, and in a number of vegetables. Attempts have been made to produce hybrid seed in wheat (Kempe et al. 2014).
Although heterosis contributes to increased yield in many crops and vegetables, the underlying biological mechanisms of heterosis are not well understood. For a long time, research has aimed to clarify this mystery. This review describes recent research on heterosis.
Traditional models of heterosis
Attempts to understand the genetic basis of heterosis began in the 1990s and several hypotheses were put forward to explain the mechanism of heterosis. All addressed levels of gene activity. The dominance hypothesis was the first to be proposed (Bruce 1910, Crow 1998, Davenport 1908, Jones 1917) and heterosis results in the suppression/complementation of deleterious recessive alleles from one parent by dominant alleles from the other parent (Fig. 1). This model predicts it would be possible to create an inbred line of equal performance to the F1 hybrid cultivar by eliminating all deleterious alleles and/or accumulating favorable alleles. The second proposal is the overdominance hypothesis, which attributes heterozygosity at individual loci as leading to heterosis (Fig. 1) (Crow 1998, East 1936, Shull 1908). The third suggestion is the epistasis hypothesis in which interactions between two or more non-allelic genes derived from the parental lines generate heterosis (Fig. 1) (Powers 1944, Richey 1942, Williams 1959). These hypotheses have been fundamental to heterosis research, but it is not clear that any one model explains the molecular mechanism of heterosis. These hypotheses are not mutually exclusive, and it is possible that heterosis is an accumulation or interaction of alleles with contributions from each mechanism.
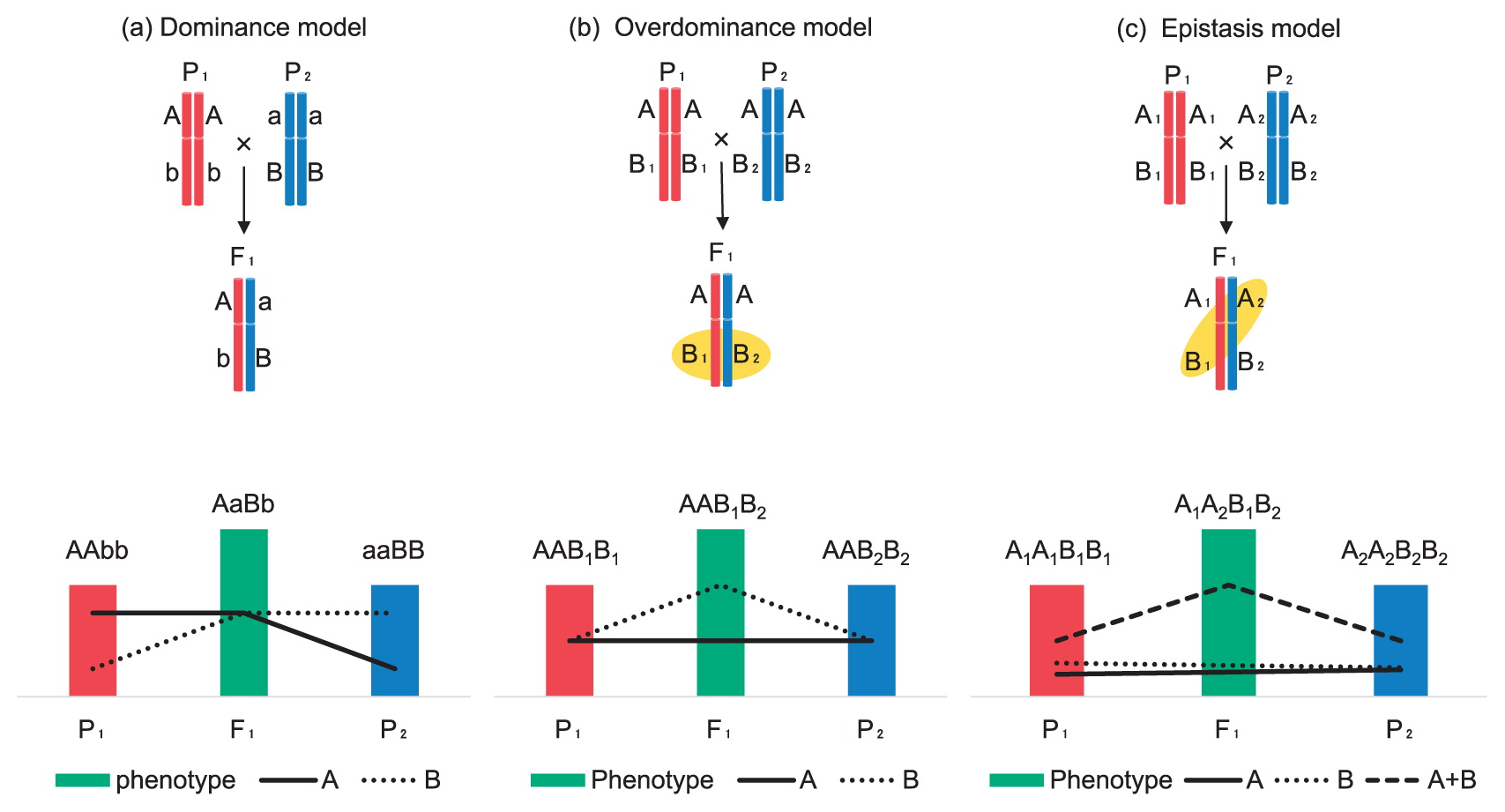
Genetic analysis of heterosis
Any positive correlation between genetic distance and heterosis has been debated (Birchler et al. 2010). Initially, in maize, it was accepted that crosses of more genetic divergent parental lines lead to greater heterosis. There was a positive correlation between genetic distance and heterosis within a range of diverged parental lines, although there was a negative correlation between genetic distance and heterosis when the genetic distance was extremely high (Moll et al. 1965). An efficient method for predicting reliable hybrid performance from parental lines is desired in F1 hybrid breeding because hybrid production is expensive, time consuming, and is labor intensive to test large number of hybrids in field trials. Recently development of DNA marker and sequence technology allows calculation of the genetic distance with high accuracy. However, positive correlation is not always observed between genetic distance and heterosis (Barth et al. 2003, Girke et al. 2012, Kawamura et al. 2016, Yang et al. 2017). Prediction of hybrid performance by genomic selection using general or specific combining ability has also been challenged (Zhao et al. 2015)
Quantitative trait loci (QTL) analysis has been performed in maize, rice, sorghum, tomato, rapeseed, and cotton in attempts to understand the genetic basis of heterosis (Lippman and Zamir 2007). Most heterosis QTL studies focus on yield-related traits in biparental populations (Lippman and Zamir 2007). Other researchers tried to identify a QTL for general or specific combining ability in hybrids using multiparental populations (Giraud et al. 2017, Zhen et al. 2017). In maize, pioneering QTL research concluded that overdominance was the main cause of heterosis (Stuber et al. 1992), and this was supported by a later study (Li et al. 2017). Other research programs led to dominance or epistasis to be considered as the main cause of heterosis in maize (Frascaroli et al. 2007, Lu et al. 2003). In rice, there is support for both the dominance hypothesis (Xiao et al. 1995) and the overdominance and epistasis hypotheses (Li et al. 2001, Luo et al. 2001). In tomato, a single heterozygous gene, SINGLE FLOWER TRUSS, contributes to fruit yield heterosis, the first example of a single overdominant gene in plants (Krieger et al. 2010). It appears that heterosis cannot be explained by any single hypothesis, and the causes of heterosis depend on the species, traits, and parental combination (Li et al. 2008).
Single nucleotide polymorphism (SNP) data of large populations have enabled a comparison of genetic architecture in a number of lines. In addition, genome-wide association studies (GWAS) using large SNP data have been incorporated into a genetic approach. In rice, genomes of 10,074 F2 lines derived from 17 hybrids were sequenced for genotyping, and QTLs for yield traits identified. Positive dominance effects tended to be overrepresented in the grain-yield-related traits, with the contribution of male alleles being larger than female alleles in most of the grain-yield QTLs (Huang et al. 2016). In winter wheat, grain yield was assessed in 1,604 hybrids and their 15 male and 120 female parental elite lines (approximately four million plants) in 11 environments, and a 90,000-SNP-array used for genotyping. Relative contributions of the genetic components of mid-parent heterosis for grain yield were estimated, and dominance, additive-by-additive, additive-by-dominance, and dominance-by-dominance variance were 16%, 50%, 21%, and 13%, respectively, suggesting that epistatic effects are greater than dominance effects in grain-yield heterosis of wheat (Jiang et al. 2017).
How additively or non-additively expressed genes can contribute to heterosis
Transcriptome analyses initially using microarray technology and later, RNA-sequencing (RNA-seq) have been used to compare parental lines with their F1 hybrids to identify genes potentially involved in heterosis.
The mode of gene action in F1 hybrids is classified as having additive and non-additive expression. Additive gene expression is where the gene expression level in F1 hybrids is equal to the average level of parental gene expression (termed mid-parent value; MPV) (Fig. 2). Non-additive gene expression occurs when the gene expression level in F1 hybrids is different from the MPV. Gene expression is further classified into high-parent dominance, low-parent dominance, over-dominance, and under-dominance (Fig. 2). High- or low-parent dominance is when expression levels in hybrids are equal to one parent but is significantly different from the second parent (Fig. 2). Over- and under-dominance is when gene expression levels in hybrids are outside the parental range (Fig. 2). In seedlings of maize, transcriptome analysis using microarrays on the parental inbred lines, B73 and Mo17, and their heterotic F1 hybrids showed all possible modes of gene action in F1 hybrids, with most genes having additive gene expression (Swanson-Wagner et al. 2006).

Transcriptome analyses have been performed for seedlings, roots, embryos, and immature ears in maize (Hu et al. 2016, Jahnke et al. 2010, Paschold et al. 2012, Stupar et al. 2008, Swanson-Wagner et al. 2006). In any tissue, there were more additively expressed genes than non-additively expressed genes, and non-additively expressed genes were tissue/stage specific. Seedlings of six F1 hybrids with a diversity of heterosis levels showed similar proportions of additively and non-additively expressed genes, suggesting that heterosis is not a consequence of the proportion of higher levels of additive or non-additive expression in F1 hybrids (Stupar et al. 2008).
In rice, transcriptome analyses have been performed for seedlings, leaves, roots, and inflorescence buds (Li et al. 2016, Song et al. 2010, Wei et al. 2009, Zhai et al. 2013). Using super-hybrid rice Lian-You-Pei-Jiu 9 (LYP9) and its parental lines, transcriptome analyses by microarray in seven tissues (leaves at seedling and tillering stages, flag leaves at booting, heading, flowering, and filling stages, and panicles at filling stages) were performed. Cluster analysis revealed transcriptome profiles of LYP9 were similar to the maternal parent in the early developmental stages but closer to the paternal parent at later stages. Non-additively expressed genes range from 0.5 to 1.4% in seven tissues, and differentially expressed genes showed tissue specificity (Wei et al. 2009). RNA-seq using young inflorescence buds at the four early developmental stages in LYP9 and its parental lines showed from 30 to 50% of genes had non-additive expression. Enriched pathways were translation, transcription, circadian rhythm, carbon fixation, and starch and sucrose metabolism (Li et al. 2016). Several genes differentially expressed between F1 hybrids and its parents were located at yield-related QTL regions, suggesting they are candidates for contributing to heterosis (Li et al. 2016). Using another super-hybrid rice Liangyou-2186 (LY2186) and its parents, transcriptome analysis by serial analysis of gene expression (SAGE) was performed in leaves at the grain-filling stage. Photosynthesis-related categories were over-represented in the differentially expressed genes, and genes involved in carbon-fixation pathway were up-regulated in F1 hybrids. The activity of enzymes involved in the carbon fixation pathways and the net photosynthetic rate were increased in the F1 hybrids, suggesting that carbon fixation is enhanced, which could be associated with heterosis (Song et al. 2010).
In B. napus shoot apical meristems and young flower buds (three stages of early flower developments), most genes showed additive expression in the F1 hybrids. Non-additively expressed genes were differentially expressed between parental lines and had high-parent dominance. These genes had over-representation of the categories related to defense and hormone responses (Shen et al. 2017).
The underlying hypothesis for a transcriptome approach is that genes whose expression changes in F1 hybrids (non-additively expressed genes) may be involved in heterosis and include genes categorized into specific pathways that may have altered regulation. The hypothesis that the mid-parent expression pattern in F1 hybrids might play a role in heterosis is considered because in many cases, the number of differentially expressed genes between parental lines was more than those between parental lines and F1 hybrids. The majority of genes showed additive gene expression, with only a small proportion of genes showing non-additive gene expression (Springer and Stupar 2007). There is no clear conclusion as to which is more important, additive or non-additive expression for generating heterosis, but both classes probably contribute.
Transcriptional changes in heterotic hybrids at the early developmental stages in A. thaliana
In F1 hybrids in A. thaliana such as C24 × Columbia-0 (Col) and C24 × Landsberg erecta (Ler), a heterosis phenotype is seen at early developmental stages with hybrids having increased cotyledon size only a few days after sowing (DAS) (Fujimoto et al. 2012a, Groszmann et al. 2014, Meyer et al. 2012). In C24 × Col hybrids as early as 3 DAS, 724 genes showed increased expression and 329 genes showed decreased transcription compared with MPV. There was a particularly high over-representation of genes with increased transcription whose products are active in the chloroplast (187 genes out of the 724) and these were mainly genes involved in chlorophyll biosynthesis and photosynthesis. The increased transcription of the chloroplast-targeted genes seen at 3 DAS was transient and was not present in cotyledons at 6 or 7 DAS. Up-regulation of the chloroplast-targeted genes was observed in the first and second leaves at 6 or 7 DAS. This up-regulation was also transient and was not present in these leaves at 10 DAS. At 10 DAS in the aerial tissue, 118 genes showed differential expression between F1 hybrids and the MPV of which 95 were up-regulated and 23 down-regulated genes. Genes categorized into response to biotic stimulus tended to be up-regulated and genes categorized into response to stress tended to be either up- or down-regulated. A small number of genes overlapped between the 3 and 10 DAS sets of differentially expressed genes. At 3 DAS, up-regulated genes showed an over-dominance mode, while non-additively expressed genes showed high-parent or low-parent dominance gene action at 10 DAS (Fujimoto et al. 2012a). Transcriptomes of Col, C24, and the reciprocal hybrids at 4, 6, and 10 DAS were also compared (Meyer et al. 2012). Seventy-five percent of differentially expressed genes showed differences in the transcript levels at only one time point, and 10% of genes were significantly different at all time points. Genes involved in defense response tended to be differentially expressed at 6 DAS, while there was no over-representation in any categories at other time points. The gene expression levels in the F1 hybrids were intermediate between the levels of parental lines or close to the level of one of the two parents, suggesting that mid-parent expression in F1 hybrids can lead to heterosis (Meyer et al. 2012). These results indicate that the non-additive gene expression profile is drastically changed in developmental stages even when they differ by only a few days (Fujimoto et al. 2012a, Meyer et al. 2012).
In C24 × Ler hybrids, transcriptome analyses have been performed on developing embryos and seedlings, and 15-day-old seedlings (Alonso-Peral et al. 2017, Groszmann et al. 2015, Shen et al. 2012, Zhu et al. 2016). In the early embryo at four days after pollination (globular stage) and six days after pollination (heart stage), the transcriptome profile of F1 hybrids was similar to the maternal line (Alonso-Peral et al. 2017). Transcriptome analyses in the early seedlings at 0, 3, 5, and 7 DAS showed the number of non-additively expressed genes at 3 DAS was more than those at the other time points, and the proportion of the over- and under-dominance gene mode at 3 DAS was higher than at the other time points. However, non-additively expressed genes at 3 DAS did not show non-additive gene expression at 5 or 7 DAS, suggesting that non-additive expression during early stages of seedling developments is mainly a consequence of differences in the timing of the gene expression pattern between F1 hybrids and parents (Zhu et al. 2016). At 0 DAS, a small number of non-additively expressed genes overlapped between reciprocal hybrids, while more non-additively expressed genes overlapped between reciprocal hybrids at 3 DAS, suggesting a weakening of the maternal effect, which is established in developing embryos from 0 to 3 DAS (Alonso-Peral et al. 2017, Zhu et al. 2016). At 3 DAS, many up-regulated genes in F1 hybrids are photosynthesis-related genes (Zhu et al. 2016), similar to the results in C24 × Col hybrids (Fujimoto et al. 2012a). In 15-day-old seedlings, differentially expressed genes tended to be down-regulated in C24 × Ler hybrids, and genes involved in flavonoid biosynthesis were down-regulated in reciprocal hybrids (Groszmann et al. 2015, Shen et al. 2012). Flavonoids inhibit auxin transport, and F1 hybrids showed faster indole-3-acetic acid (IAA) transport than parental lines, suggesting that heterosis is at least partially due to increased auxin signaling (Shen et al. 2012). Genes involved in salicylic acid (SA) biosynthesis were down-regulated in C24 × Ler and C24 × Col hybrids in 15-day-old seedlings and a decrease in SA concentration in C24 × Ler compared with MPV was observed, suggesting that a reduction in SA concentration and associated changes in gene expression levels in the C24 × Ler hybrids contribute to heterosis (Groszmann et al. 2015).
Photosynthetic capacity is important for heterosis
In A. thaliana, F1 hybrids between C24 and Col and between C24 and Ler showed earlier up-regulation of chloroplast-targeted genes in 3-day-old cotyledons (Fujimoto et al. 2012a, Zhu et al. 2016). Genes involved in photosynthesis-related categories were also up-regulated in leaves of super-hybrid rice LY2186 at the grain-filling stage (Song et al. 2010). A commercial F1 hybrid cultivar of Chinese cabbage (Brassica rapa var. pekinensis), W39, showed vegetative heterosis at early developmental stages and yield heterosis (25% greater than the better parent) (Saeki et al. 2016). Increased cotyledon area in W39 was obvious from 2 to 4 DAS, and genes involved in chloroplast-targeted genes were up-regulated at 2 DAS in W39. As in A. thaliana, this up-regulation was transient (Saeki et al. 2016). In the C24 × Col hybrids, transient up-regulation of chloroplast-targeted genes did not lead to increased photosynthesis activity per unit area or chlorophyll content per unit fresh weight (Fujimoto et al. 2012a), while super-hybrid rice LY2186 showed increased activity of enzymes involved in carbon fixation pathways and net photosynthetic rate (Song et al. 2010). In the heterotic hybrid, Tentaka, in Sorghum bicolor, the CO2 assimilation rate per leaf area did not differ from its parental lines (Tazoe et al. 2016). In all cases, the amount of photosynthesis per individual plant in F1 hybrids is larger than that of parental lines because of increased leaf area in heterotic hybrids.
The relationship between transient up-regulation of chloroplast-targeted genes and increased leaf area is not clear. In A. thaliana, cell size and chloroplast numbers per cell were correlated (Fujimoto et al. 2012a, Pyke and Leech 1991), and photosystem II chlorophyll a/b-binding polypeptide gene (Lhcb) transcription is coordinated with cell size (Meehan et al. 1996). When the leaf area is increased in F1 hybrids, cell number and/or cell size increased with increased chloroplast number or chlorophyll content, suggesting that up-regulation of chloroplast-targeted genes might be involved in this step. To confirm the importance of a relationship between chlorophyll biosynthesis/photosynthesis and heterotic biomass, norflurazon, an inhibitor of phytoene desaturase and hence chlorophyll synthesis and photosynthesis, was applied. Treatment of seedlings at 3 DAS with norflurazon resulted in the heterotic response of the hybrid being absent at 21 DAS in A. thaliana (Fujimoto et al. 2012a). In Chinese cabbage, seeds were grown on Murashige and Skoog (MS) medium for one week, and transferred to MS medium with 1 μM norflurazon and grown a further two weeks. In this case, heterosis was obvious in the first and second leaves of the W39 hybrid in spite of leaves being without chlorophyll. In contrast, seeds grown on MS medium with 1 μM norflurazon for one week and transferred to MS medium without norflurazon for three weeks did not show any heterosis in the third and fourth leaves, which were green (Fig. 3) (Saeki et al. 2016). These results suggest that chlorophyll biosynthesis or photosynthesis in the first week of cotyledon growth is important for increased leaf size in F1 hybrids and even after the cotyledon stage. The up-regulation of chloroplast-targeted genes in the cotyledon and photosynthesis at the cotyledon stage are important for increased leaf area in F1 hybrids, and this increased leaf area could lead to an increased amount of photosynthesis per individual plant, leading to yield heterosis (Saeki et al. 2016).
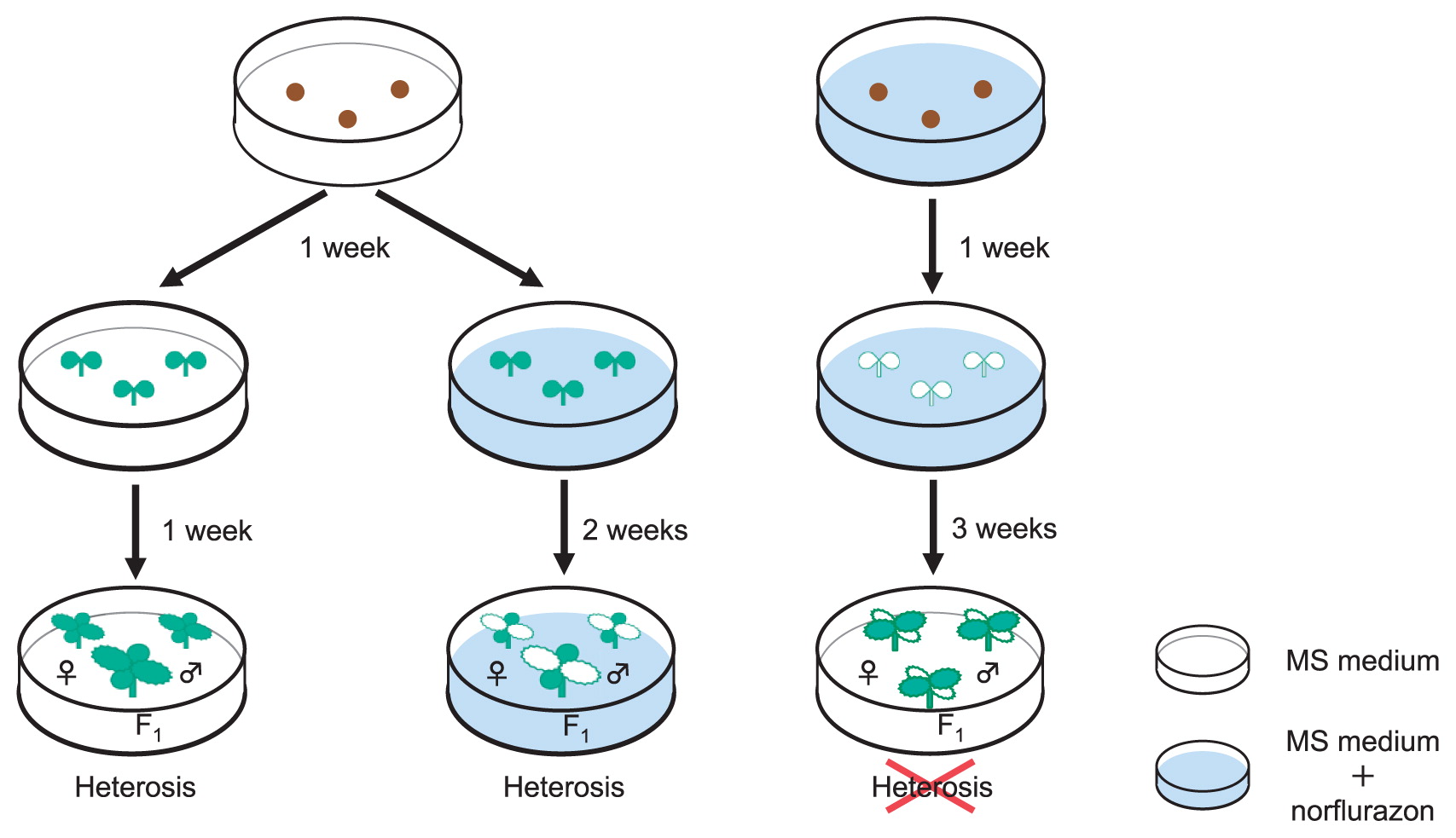
Is circadian-mediated gene expression involved in heterosis?
The possibility that heterosis is caused by a change of gene expression of master genes affecting downstream gene expression has been discussed. Altered expression of the central clock genes in the F1 increases expression levels of downstream genes involved in energy and metabolism including photosynthesis and starch pathways, and could lead to biomass heterosis (Ni et al. 2009).
Inter-specific hybrids show non-additive gene expression and a hybrid vigor-like phenotype (Fujimoto et al. 2011, Osabe et al. 2012a, Tonosaki et al. 2016). The progeny of allotetraploids synthesized by inter-specific hybridization between A. thaliana and A. arenosa showed growth vigor and non-additive expression of over 1,400 genes. Of these non-additively expressed genes, many genes involved in energy and metabolism pathways are up-regulated. Some genes contain at least one CIRCADIAN CLOCK ASSOCIATED 1 (CCA1) binding site or evening element. CCA1 and LATE ELONGATED HYPOCOTYL (LHY) expression levels in allotetraploids were lower than the MPV at Zeitgeber time (ZT) 6–12 and higher than the MPV at ZT15 (ZT reflects the hours since dawn). TIMING OF CAB EXPRESSION 1 and GIGANTEA expression were inversely correlated with CCA1 and LHY expression. These results indicate that amplitude but not the phase of the circadian clock is altered in allotetraploids, suggesting that this change leads to up-regulation of genes in chlorophyll and starch metabolic pathways and an increased amount of chlorophyll and starch. Similar results were observed in an intra-specific hybrid between C24 and Col accessions of A. thaliana, suggesting that altered expression of circadian oscillator genes is associated with heterosis in C24 × Col hybrids as well as in allotetraploids. Ni et al. (2009) showed 12% more total chlorophyll and 10% more starch in C24 × Col hybrids than the higher parent. By contrast, chlorophyll content per unit fresh weight and photosynthetic activity per unit area in C24 × Col hybrids was not increased (Fujimoto et al. 2012a). The increased expression of genes reported by Ni et al. (2009) was not apparent in the transcriptome analyses by two other groups using the same C24 × Col hybrids (Fujimoto et al. 2012a, Meyer et al. 2012). As the non-additively expressed genes are stage-specific (Fujimoto et al. 2012a, Meyer et al. 2012), there is a possibility that a difference in developmental timing is responsible for the differences reported in the transcriptomes.
In the hybrid between C24 and Ler, expression levels of CCA1 were 20% to 30% lower than the MPV at ZT6 and CCA1 expression peaks in C24 × Ler were lower than Ler × C24 hybrids at ZT6 (e.g. ZT6 is six hours from dawn). This difference of CCA1 expression between reciprocal hybrids was correlated with difference of starch accumulation; lower CCA1 expression at ZT6 correlates with increased biomass. The DNA methylation level at CHH sites (H is A, T, or C) in the promoter region of CCA1 in Ler × C24 hybrids was higher than that in C24 × Ler hybrids, suggesting there was negative correlation between DNA methylation level and CCA1 expression. In addition, Ler × argonaute 4 (ago4) (Ler) hybrids showed lower CCA1 expression peaks, higher CHH methylation levels in the promoter region of CCA1, and greater dry weight than those in ago4 (Ler) × Ler hybrids. These results suggest that parent-of-origin effect on CHH methylation via RNA-directed DNA methylation (RdDM) affects CCA1 expression amplitude and leads to growth difference between reciprocal hybrids (Ng et al. 2014). It is not clear how this model is related to the heterosis shown in both reciprocal hybrids (C24 × Ler or Ler × C24 hybrids).
Unlike A. thaliana, in maize, there is a temporal shift of ZmCCA1-binding to targets to the early morning; the proportion of ZmCCA1-binding peaks at ZT3 was higher than at ZT9 or ZT15 in hybrids but there was no difference between ZT3 and ZT9 in parental lines. The target genes of this temporal shift to the early morning showed up-regulation in the morning in hybrids compared with parental lines. From these results, a phase-shift model for heterosis was predicted; the altered temporal binding of ZmCCA1 to photosynthetic and metabolic genes causes non-additive gene expression levels and increased carbon fixation capacity, leading to heterosis (Ko et al. 2016).
It is certain that circadian rhythm is important for the growth of plants (Ni et al. 2009), but further research will be necessary to determine whether an alteration of the amplitude or phase-shift of the circadian rhythm directly up-regulates the expression of genes involved in chlorophyll and starch metabolic pathways and increases the amount of chlorophyll or starch in heterotic hybrids.
Inter- and Intra-specific hybridization cause non-additive small RNA expression
Small RNAs are involved in a broad range of function; micro RNAs (miRNAs) are predominantly 21 nucleotides (nt) in length and involved in plant development (Cui et al. 2017, Guo et al. 2016). Small interfering RNA (siRNAs) are predominantly 24 nt in length and involved in heterochromatin maintenance and silencing of transposable elements (TEs) (Wendte and Pikaard 2017). 24-nt siRNAs are involved in the process of de novo DNA methylation, termed RdDM. Two plant specific RNA polymerases, RNA Polymerase IV (Pol IV) and Pol V, along with RNA-DEPENDENT RNA POLYMERASE 2 (RDR2), DICER-LIKE 3 (DCL3), and AGO4 all function in this RdDM pathway in A. thaliana (Fig. 4) (Wendte and Pikaard 2017). Pol IV and RDR2 are involved in the biogenesis of 24-nt siRNAs and are physically associated in both A. thaliana and maize (Haag et al. 2012, 2014, Law et al. 2011).

Several reports have examined global small RNA profiles using heterotic F1 hybrids and their parental lines in maize, rice, and A. thaliana (Barber et al. 2012, Groszmann et al. 2011a, Ha et al. 2009, He et al. 2010, Kenan-Eichler et al. 2011, Li et al. 2012, Shen et al. 2012). The first report comparing small RNA profiles between the hybrids and parents used inter-specific hybrids between A. thaliana and A. arenosa, which display growth vigor. siRNAs were reduced in the F1, but their levels recovered in the F7 (Ha et al. 2009). In another case of inter-specific hybrids between tetraploid Triticum turgidum ssp. durum (AABB genome) and the diploid Aegilops tauschii (DD genome), siRNAs were not changed in hybrids (ABD genome), but were decreased in allopolyploids (AABBDD genome) (Kenan-Eichler et al. 2011). The inter-specific hybrid of a different parental combination, T. turgidum ssp. dicoccon (AABB) and Ae. tauschii ssp. strangulata (DD), was also used for small RNA profiles in three types of tissues, 7-day-old seedling, heading-stage young spikes, and immature seed at 11 days after flowering. In all three tissues, the levels of 24-nt siRNAs in Ae. tauschii were smaller than those in T. turgidum, and in allohexaploid progeny levels appeared to be similar to those in T. turgidum (Li et al. 2014). These results indicate changes of expression of 24-nt siRNA occur in some tissues of the allopolyploids compared with the progenitor species.
In the case of intra-specific hybrids, 24-nt siRNAs tended to be down-regulated in heterotic hybrids compared with their parental lines. In rice heterotic hybrid between ‘Nipponbare’ and 93-11, siRNAs were down-regulated in the hybrid and most showed a low parent pattern (He et al. 2010), although this conclusion has been changed following re-analysis of the data by the same group; only a small fraction of siRNAs (<0.8%) changed between hybrid and parental lines (Shen et al. 2012). In B. napus, the number of 24-nt siRNA clusters in F1 hybrids was higher than the MPV in the shoot apical meristems or young flower buds (Shen et al. 2017). The heterotic hybrid of A. thaliana, C24 × Ler, showed a reduction of 24-nt siRNAs relative to the parental lines at loci where the parents had different 24-nt siRNAs levels (Groszmann et al. 2011a). Another research group showed that most siRNA clusters (>93%) were not differentially expressed between parents and hybrids, though differentially expressed siRNA clusters tended to be at low parent value (Shen et al. 2012). The global small RNA profile has been examined using another combination of A. thaliana accessions, Ler and Col, which showed biomass heterosis in short day conditions. Sixty percent of siRNA clusters showed non-additive expression and 80% were at low parent values (Li et al. 2012). In the heterotic maize hybrid between B73 and Mo17, siRNA clusters were additive in the shoot apex, while siRNA clusters in the ear showed larger deviations, especially below mid-parent levels (Barber et al. 2012).
Global or local reductions in 24-nt siRNAs in hybrids may be a universal phenomenon but whether the reduction of 24-nt siRNAs is responsible for heterosis is not clear. 24-nt siRNAs are involved in RdDM and it is generally considered that RdDM can lead to transcriptional repression not only in TEs but also in protein-coding genes through DNA methylation; reduction of 24-nt siRNAs may result in changes of DNA methylation levels leading to change of expression level in TEs and some protein-coding genes. As siRNAs have the ability to modify epigenetic marks in trans, siRNA derived from one parental allele can modify the epigenetic state in the other parental allele in hybrids (Greaves et al. 2015, Groszmann et al. 2013). The number of re-activated transposons in an RdDM defective mutant was much smaller than in the hypomethylated decrease in DNA methylation 1 (ddm1) mutant (Zemach et al. 2013), suggesting that the effect of silencing of transposons by RdDM is not very large; siRNA-targeted TEs are associated with reduced gene expression in both A. thaliana and A. lyrata (Hollister et al. 2011). In C24 × Ler hybrids, decreased 24-nt siRNAs are largely associated with flanking regions of protein-coding genes and there was a correlation between change of siRNA levels with DNA methylation and expression levels at several loci (Groszmann et al. 2011a).
In the rdr2 mutant, which has defects in 24-nt siRNA biogenesis, only 70 genes had differential expression compared with wild type (wt) plants, and 26 of these 70 genes had siRNAs in their flanking regions (Kurihara et al. 2008), indicating RdDM has only a small impact on expression of protein-coding genes. To clarify the relationship between siRNAs and heterosis, mutants in biogenesis of 24-nt siRNAs were analyzed in maize and A. thaliana (Barber et al. 2012, Kawanabe et al. 2016, Zhang et al. 2016a). Heterosis was fully maintained in maize hybrids, homozygous for mediator of paramutation 1 (mop1), the ortholog of RDR2 (Barber et al. 2012). The heterosis phenotype is not affected in hybrids between Col and C24 accessions in A. thaliana with homozygous mutations in either rdr2 or nuclear RNA polymerase IV, subunit 1 (nrpd1; largest subunits of Pol IV), which are key genes involved in biogenesis of 24-nt siRNAs (Kawanabe et al. 2016, Zhang et al. 2016a, 2016b). Hybrids having mutations in ago4, ago6, or nuclear RNA polymerase V, subunit 1 (nrpe1; largest subunit of Pol V), which are also components of RdDM, maintained heterosis (Zhang et al. 2016b). Hybrids having mutations in defective in meristem silencing 3 (dms3), defective in RNA-directed DNA methylation 1 (drd1), or RNA directed DNA methylation 1 (rdm1), which are components of the DRD1-DMS3-RDM1 (DDR) complex involved in Pol V function, also maintained heterosis (Zhang et al. 2016b). These results indicate that the RdDM pathway is not required for the establishment of heterosis in maize or A. thaliana.
Is DNA methylation a key factor in heterosis?
A comparison of DNA methylation states between parental lines and their progenies generated from single seed descent over 30 generations has shown that the rate of spontaneous changes of DNA methylation is higher than the rate of spontaneous genetic mutations (Becker et al. 2011, Ossowski et al. 2010, Schmitz et al. 2011), suggesting that sequence-independent epialleles play important roles in phenotypic diversity (Becker et al. 2011, Fujimoto et al. 2012b, Schmitz et al. 2011). DNA methylation, which is one epigenetic modification, is classified into two types, de novo (see above) and maintenance DNA methylation. In maintenance DNA methylation, CG context methylation is maintained by METHYLTRANSFERASE 1 (MET1), and non-CG contexts are maintained by DOMAINS REARRANGED METHYLTRANSFERASE 2 (DRM2), CHROMOMETHYLASE 2 (CMT2), and CMT3 (Osabe et al. 2012b, Zemach et al. 2013). In addition to DNA methyltransferases, a chromatin remodeling factor, DDM1, is involved in the maintenance of DNA methylation (Fig. 4) (Fujimoto et al. 2008, Sasaki et al. 2011, Vongs et al. 1993, Zemach et al. 2013).
To identify loci with epigenetic regulation causing phenotypic variation, populations of epigenetic recombinant inbred lines (epi-RILs) between parents, which differed only in epigenetic marks, such as hybrids between met1 and wt Col, or between ddm1 and wt Col have been generated in A. thaliana (Johannes et al. 2009, Reinders et al. 2009, Teixeira et al. 2009). Stable inheritance of complex traits such as flowering time, primary root length, plant height, and biomass has been observed in these epi-RIL populations, providing evidence that epigenetic variation can contribute to complex traits (Cortijo et al. 2014, Johannes et al. 2009, Reinders et al. 2009). Enhanced growth similar to heterosis was observed in several of the hybrids between wt and specific epiRIL lines. In the case of epiRIL lines derived from hybrids between met1 and wt, 34 selected epiRILs were crossed with wt Col, and rosette sizes in F1 hybrids were examined. Twenty-eight of 34 hybrids did not exceed the better parent. Two F1 hybrids had enlarged rosettes at a young stage but not after 14 days. The remaining four F1 hybrids showed increased rosette size until bolting, and the F1 hybrid between one epiRIL, epi31, and wt showed consistent vigorous growth. The epi31 × wt hybrids showed this vigorous growth, but wt × epi31 did not, suggesting that parent-of-origin effects influence hybrid growth (Dapp et al. 2015). This research supports the involvement of epigenetic regulation and interactions in heterosis (Dapp et al. 2015). In the case of other epiRIL lines derived from the hybrids between ddm1 and wt, the F1 hybrids between Col and 19 epiRILs were generated, and three F1 hybrids showed greater than 15% increased leaf area compared with the better parent, suggesting that heterosis in F1 hybrids generated from epigenetically divergent lines may be a general phenomenon (Lauss et al. 2016).
MutS HOMOLOG1 (MSH1) encodes a protein dually targeted to mitochondria and plastids and is involved in organelle genome stability (Abdelnoor et al. 2003, Xu et al. 2011). Disruption of MSH1 causes plant phenotypes such as leaf variegation, dwarfism, altered leaf structure, and late-flowering in A. thaliana. An altered phenotype caused by disruption of MSH1 was also observed in other species such as sorghum, tomato, and soybean (Xu et al. 2012). Disruption of MSH1 causes changes of DNA methylation in A. thaliana (Virdi et al. 2015). Enhanced vigor was observed in F4 generations derived from the hybrids between wt and msh1 in A. thaliana (Virdi et al. 2015). MSH1-RNA interference (RNAi) suppression in sorghum or tomato showed altered phenotypes, which were inherited even in transgene-null offspring. Crossing between wt and transgene-null plants with developmental alterations results in enhanced heritable growth vigor in the progeny lines in sorghum and tomato (de la Rosa Santamaria et al. 2014, Yang et al. 2015b). These results suggest that epigenetic reprogramming can result in enhanced growth patterns; this may lead to its use in epigenetic breeding in crop plants (Virdi et al. 2015, Yang et al. 2015b).
Recent progress has pointed to the possibility that epigenetic regulation might contribute to heterosis (Greaves et al. 2015, Groszmann et al. 2011b, 2013, Springer 2013). DNA methylation states have been compared between heterotic F1 hybrids and parental lines in rice, B. napus, and A. thaliana (Chodavarapu et al. 2012, Greaves et al. 2012, He et al. 2010, Shen et al. 2012, 2017, Zhang et al. 2016a). In B. napus, F1 hybrids had a slightly higher level of DNA methylation at all three sequence contexts, CG, CHG, and CHH sites (H is A, T, or C). The number of unmethylated cytosines in F1 hybrids was decreased, while the number of highly methylated cytosines was not increased. As increased 24-nt siRNA clusters were observed in F1 hybrids, the authors suggested that increased 24-nt siRNAs are directly correlated with increased DNA methylation levels (Shen et al. 2017). In rice using fully expanded leaves from six-week-old plants in ‘Nipponbare’ and indica 93-11, and their reciprocal hybrids, DNA methylation levels in the F1 hybrids were intermediate to parental methylation levels. Differential methylation was strongly associated with regions where siRNA production was unequal in the two parents (Chodavarapu et al. 2012).
In the case of A. thaliana using 15-day-old seedlings of C24, Ler, and its reciprocal hybrids, increased DNA methylation levels at all three contexts in both reciprocal hybrids relative to parental lines were observed. F1 hybrids had increased DNA methylation relative to parental lines in both protein-coding genes and TEs, and the increase in TEs was greater than in protein-coding genes. In addition, increased DNA methylation in F1 hybrids occurred predominantly in regions covering siRNA clusters, especially the regions that were differentially methylated between the parents (Shen et al. 2012). In the case of this parental combination using the immature floral buds, F1 hybrids showed differences of DNA methylation compared with parental lines, especially the regions differentially methylated between the parental lines. Increases in DNA methylation were more frequent than decreases in F1 hybrids (Greaves et al. 2012). Change of DNA methylation states in the F1 hybrids results from trans-chromosomal methylation (TCM) (an increase in methylation at a locus with a previously low methylation allele gaining methylation to resemble the more heavily methylated allele) or trans-chromosomal demethylation (TCdM) (loss of methylation at a genomic segment) (Fig. 5), and TCM and TCdM events in hybrids are largely dependent on 24-nt siRNAs (Greaves et al. 2012). In the case of A. thaliana using 14-day-old seedlings of C24, Col and their reciprocal hybrids, increased DNA methylation levels in hybrids were observed in all three contexts. Regions differentially methylated by TCM and TCdM were observed in F1 hybrids and 24-nt siRNAs were associated with them.
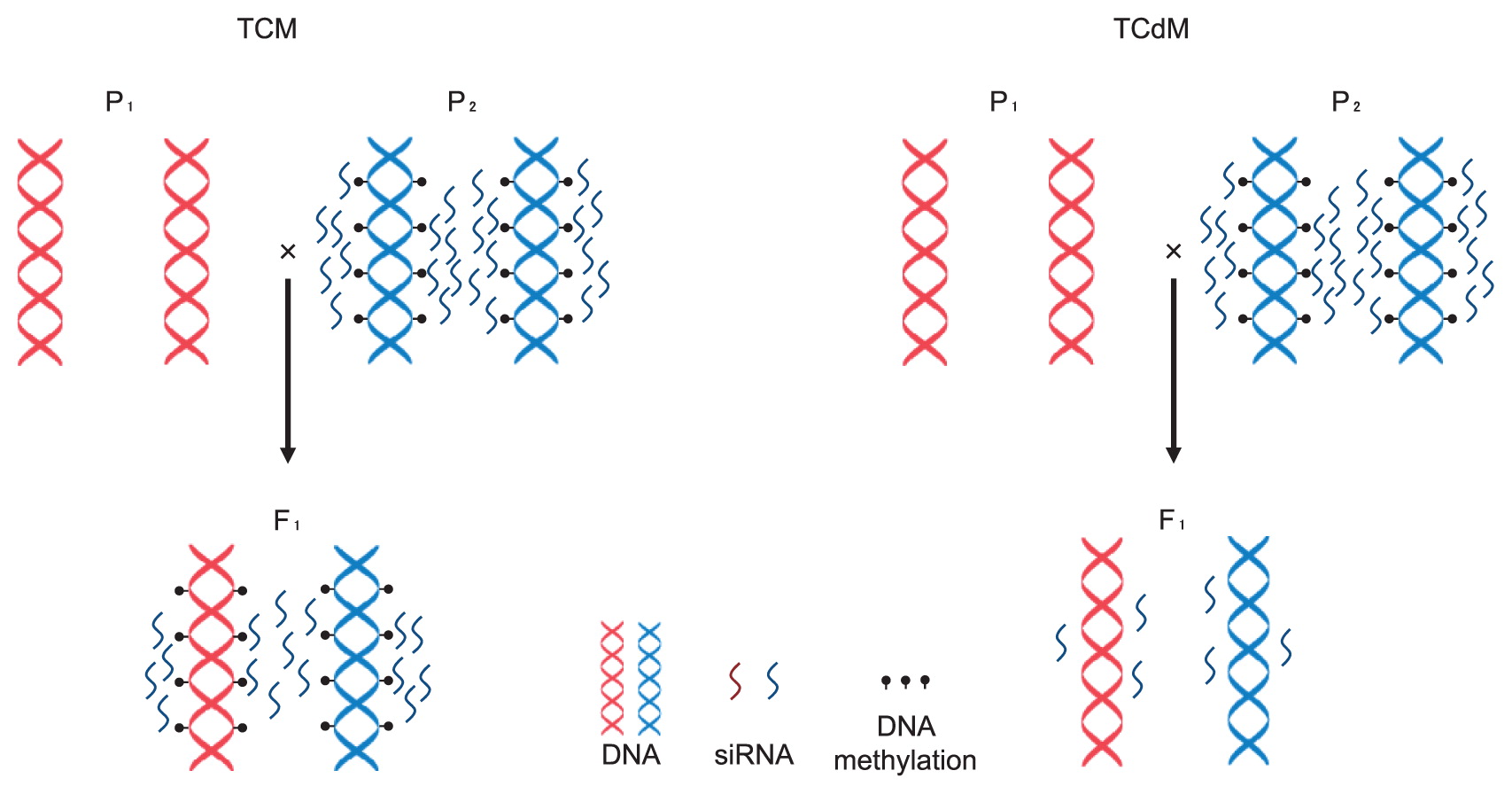
In the hybrids having mutations in both nrpd1 (pol iv mutant) and nrpe1 (pol v mutant), TCM and TCdM were abolished, but hybrids maintained the heterosis phenotype, suggesting that these DNA methylation interactions are not critical for heterosis (Zhang et al. 2016a). More than 10,000 regions of non-additively inherited DNA methylation in epihybrids occur between met1 and wt, though these F1 plants do not show superior performance, suggesting that creation of new non-parental epialleles in F1 hybrids is independent of heterosis (Rigal et al. 2016).
Recently two groups showed that DDM1 is a major regulator of heterosis (Kawanabe et al. 2016, Zhang et al. 2016b). The F1 hybrids having homozygous mutations in ddm1 had reduced vegetative heterosis in C24 × Col hybrids (Kawanabe et al. 2016, Zhang et al. 2016b). The reduction of vegetative heterosis by defects in DDM1 function was also observed in the C24 × Cvi and the Col × Cvi hybrid, the level of reduction of heterosis being dependent on the combination of parental lines (Kawanabe et al. 2016). In the hybrid between a heterozygous ddm1-9 mutant in C24 and a homozygous ddm1-1 mutant in Col, plants homozygous for the ddm1 mutation were smaller than heterozygotes. However, some plants homozygous for ddm1 showed heterosis as great as the plants having a heterozygous ddm1 mutation, and some plants with heterozygous ddm1 reduced heterosis to the level in the homozygous ddm1 hybrid plants (Kawanabe et al. 2016). In both cases there was an identical genetic background except for the ddm1 mutation. These effects may result from the previous methylation state of the genome in the ddm1 parent, and genes or chromosomal segments important for heterosis coming from the ddm1 parent having an altered level of DNA methylation.
To examine the involvement of MET1 in heterosis, two approaches were tested; using met1-RNAi knockdown lines in a C24 background or BC2F1 lines obtained by backcrossing of C24 to a hybrid with a met1 mutant of Col. Hybrids between met1-RNAi (C24) and met1-1 (Col) or BC2F1 (C24) × met1-1 (Col) hybrid with homozygous met1 did not show reduced level of heterosis, indicating that MET1-dependent maintenance of CG methylation is not involved in heterosis in C24 × Col hybrids (Kawanabe et al. 2016). An F1 hybrid between hypomethylated plants was developed by crossing ddm1-9 in C24 and met1-1 in Col. The ddm1-9 × met1-1 hybrid (DDM1/ddm1, MET1/met1) showed heterosis levels like the wild type C24 × Col hybrids, indicating that a hypomethylated state of parental lines does not cause a decreased heterosis phenotype (Kawanabe et al. 2016).
The results from vigorous growth in F1 hybrids between epiRILs and wt or reduction of heterosis levels by disruption of DDM1 function, which is involved in maintenance of DNA methylation in all three sequence contexts, suggests that alterations in DNA methylation affect the level of heterosis. However, the mechanism is unclear and further study will be required to understand how DDM1 regulates heterosis.
Heterosis and stress tolerance
Most heterosis research has been focused on growth vigor or increased yield, and there is little research on heterosis and stress tolerance. However, if stress tolerance and yield heterosis could be combined, these hybrids could be excellent cultivars.
The relationship between biomass heterosis and change of gene expression in stress response has been investigated. Transcriptomes of three-week-old seedlings at ZT0, ZT6, and ZT15 were compared between a C24 × Col hybrid and its parental lines. Genes categorized into photosynthetic and stress-responsive pathway tended to show differential expression between F1 and MPV. At ZT6, genes involved in the photosynthetic pathway were mostly up-regulated in F1 hybrids, while genes involved in abiotic or biotic stress tended to be down-regulated. Using the ACCELERATED CELL DEATH 6 (ACD6) or COLD REGULATED 78 (COR78) gene as a biotic or abiotic stress marker gene, respectively, gene expression levels of these two genes at ZT0, ZT9, and ZT18 were examined in ten accessions of A. thaliana. Ten accessions were divided into two groups, those where ACD6 genes showed a higher expression level at ZT18 and those where COR78 genes showed a higher expression level at ZT6. The biomass heterosis was correlated with difference of expression levels of ACD6 at ZT18 or COR78 at ZT9 between parental lines, suggesting that biomass heterosis is dependent on larger expression differences in stress-responsive genes between parental lines (Miller et al. 2015). A model of balancing stress response to promote biomass heterosis has been proposed (Miller et al. 2015). Hybrid necrosis, which causes slow growth, wilting, discoloration, and lethality in intra- and inter-specific hybrids, would result from an autoimmune-like response through epistatic interaction between resistance genes (Tonosaki et al. 2016). In F1 hybrids, heterosis is opposite to necrosis in plant growth, suggesting that the repression of genes related to biotic stress may result in heterosis because necrosis is caused by induction of biotic stress genes. In some plant species, genes involved in biotic stress in F1 hybrids with growth vigor tended to be down-regulated compared with those in their parental lines, and there could be a negative trade-off between expression of defense response genes and growth heterosis (Fig. 6) (Miller et al. 2015).
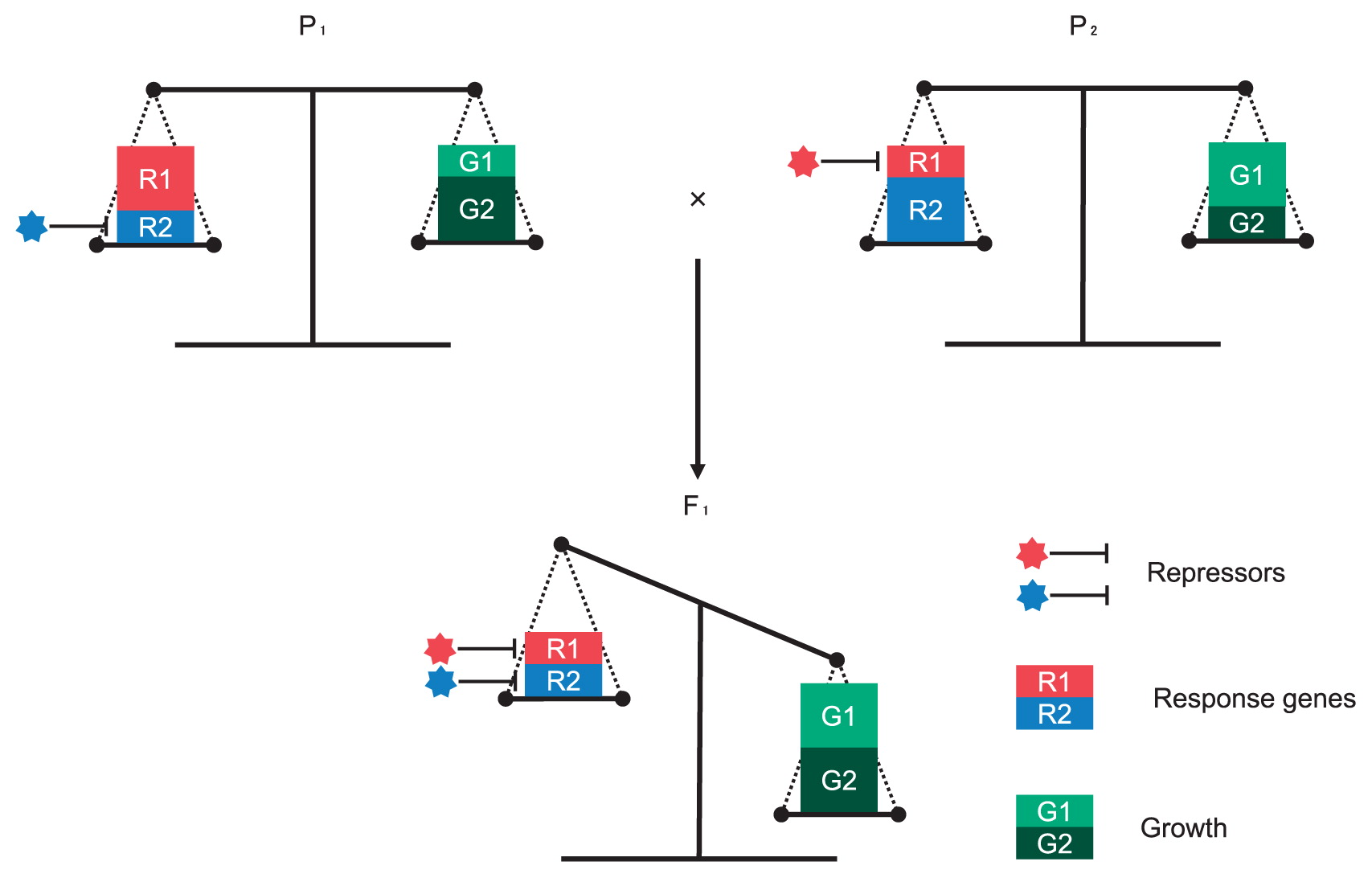
In the F1 hybrids between C24 and Ler, reduction of the expression levels in defense response genes was confirmed, but hybrids did not show greater susceptibility to P. syringae in F1 hybrids than in its parental lines (Groszmann et al. 2015). In other combinations of parental lines in A. thaliana, there was no relationship between change of gene expression in biotic stress pathways and biomass heterosis (Yang et al. 2015a). In the case of Fusarium head blight (FHB) resistance in wheat, 1,604 hybrids and 120 female and 15 male parental lines were evaluated for FHB resistance and heterosis levels. The mean better parent heterosis of FHB resistance was 2% and there was no correlation between FHB resistance and plant height, indicating growth heterosis is independent of FHB resistance (Miedaner et al. 2017). In the case of Fusarium yellows resistance gene in Chinese cabbage, resistance was largely dependent on the resistance gene itself as generally thought (Pu et al. 2012, Shimizu et al. 2014, 2015), and the growth vigor does not contribute to Fusarium yellows resistance (Kawamura et al. 2015, 2016, Saeki et al. 2016).
Though there is little evidence of a positive relationship between biomass heterosis and stress tolerance, there is some evidence of heterosis in stress tolerance. In A. thaliana, heterosis in disease resistance against P. syringae pv. tomato (Pst) DC3000 was examined using F1 hybrids between Col and 20 accessions. Two F1 hybrids (Col × Ler, Col × Sei-0) showed heterosis in Pst DC3000 resistance, while the other 18 F1 hybrids did not have any heterosis. The Col × Sei-0 hybrids showed up-regulation of genes involved in the SA biosynthesis pathway compared with parental lines, contrasting to Col × Aa-0 hybrids, which did not show heterosis in disease resistance. In addition, Col × Sei-0 hybrids accumulated more SA than parental lines at one day post-inoculation, but Col × Aa-0 hybrids did not. PHYTOALEXIN DEFICIENT 4 (PAD4) functions upstream of the SA biogenesis pathway, and the Col × Ler hybrids with homozygous pad4 mutation did not show heterosis in disease resistance, indicating that increased levels of SA resulting from enhanced activation of the SA biosynthesis pathway in hybrids are important for heterosis in disease resistance (Yang et al. 2015a).
Heterosis in freezing tolerance in reciprocal F1 hybrids between C24 and Col accessions was observed in both acclimated and non-acclimated plants. The content of soluble sugars such as fructose, glucose, raffinose, and sucrose in leaves was increased more in the F1 hybrids than the parental lines in the acclimated state. Expression levels of some cold-regulated genes were examined after three hours or 14 days of cold treatment, but all genes in F1 hybrids showed expression levels within the parental range (Rohde et al. 2004). Further combinations of A. thaliana accessions were tested and heterosis in freezing tolerance was observed in some F1 hybrids. Heterosis was observed more frequently in crosses involving C24 than in crosses involving Col. There was a moderate correlation between heterosis effects in freezing tolerance and sugar contents or flavonol content (Korn et al. 2008). In addition, metabolic profiles in five parental accessions and eight F1 hybrids in A. thaliana revealed that the pathway leading to raffinose or tricarboxylic acid cycle intermediates can be used to predict heterosis in freezing tolerance (Korn et al. 2010).
The data are not extensive but overall there was no direct relationship between biomass heterosis and heterosis in freezing tolerance or disease resistance. However, it may be possible to produce better cultivars if the biomass heterosis and the stress tolerance heterosis are combined.
Perspective
Plant breeders have been very successful in increasing crop yields and the phenomenon of heterosis has contributed to these advances for about 100 years. However, global food shortage due to population increase and global climate change is imminent, so further increases of crop production are critical. While heterosis has already contributed to increased yield, we do not understand the molecular mechanisms of heterosis. Recently, the advent of high-throughput sequencing has enabled the comparison of levels of gene expression between hybrids and their parents and identification of metabolic pathways altered in hybrids. Photosynthesis and chloroplast-targeted genes are up-regulated in hybrids relative to parents suggesting earlier development in heterotic hybrids (Fig. 7) (Fujimoto et al. 2012a, Saeki et al. 2016, Zhu et al. 2016). High throughput sequencing has also enabled dissection of the parental allelic contributions to gene expression and epigenetic states (Chodavarapu et al. 2012, Saeki et al. 2016). The chromatin remodeler DDM1 appears essential for the development of full heterosis (Fig. 7) (Kawanabe et al. 2016, Zhang et al. 2016b). Fixing heterosis so that it continues beyond the F1 generation is a challenging strategy for increasing yield, and “Hybrid Mimics”, which are pure breeding lines having the characteristics of hybrids and fix heterosis in subsequent generations, would be an option (Wang et al. 2015, 2017). Combining heterosis in different characters such as yield heterosis and stress tolerance heterosis could enhance yield. Research on the big issue of heterosis should continue as a part of enhancing food production.

Acknowledgments
This work was supported in part by an Open Partnership Joint Projects of JSPS Bilateral Joint Research Projects (14544567), Scientific Research on Innovative Areas (24113509), Grant-in-Aid Young Scientists (A) (15H05614) (JSPS), and PREST (JPMJPR12B8) (JST) to R. Fujimoto.
Literature Cited
- Abdelnoor, R.V., R. Yule, A. Elo, A.C. Christensen, G. Meyer-Gauen and S.A. Mackenzie (2003) Substoichiometric shifting in the plant mitochondrial genome is influenced by a gene homologous to MutS. Proc. Natl. Acad. Sci. USA 100: 5968–5973.
- Alonso-Peral, M.M., M. Trigueros, B. Sherman, H. Ying, J.M. Taylor, W.J. Peacock and E.S. Dennis (2017) Patterns of gene expression in developing embryos of Arabidopsis hybrids. Plant J. 89: 927–939.
- Barber, W.T., W. Zhang, H. Win, K.K. Varala, J.E. Dorweiler, M.E. Hudson and S.P. Moose (2012) Repeat associated small RNAs vary among parents and following hybridization in maize. Proc. Natl. Acad. Sci. USA 109: 10444–10449.
- Barth, S., A.K. Busimi, H. Friedrich Utz and A.E. Melchinger (2003) Heterosis for biomass yield and related traits in five hybrids of Arabidopsis thaliana L. Heynh. Heredity (Edinb) 91: 36–42.
- Becker, C., J. Hagmann, J. Müller, D. Koenig, O. Stegle, K. Borgwardt and D. Weigel (2011) Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 480: 245–249.
- Birchler, J.A., H. Yao, S. Chudalayandi, D. Vaiman and R.A. Veitia (2010) Heterosis. Plant Cell 22: 2105–2112.
- Bruce, A.B. (1910) The mendelian theory of heredity and the augmentation of vigor. Science 32: 627–628.
- Cheng, S.H., J.Y. Zhuang, Y.Y. Fan, J.H. Du and L.Y. Cao (2007) Progress in research and development on hybrid rice: a super-domesticate in China. Ann. Bot. 100: 959–966.
- Chodavarapu, R.K., S. Feng, B. Ding, S.A. Simon, D. Lopez, Y. Jia, G.L. Wang, B.C. Meyers, S.E. Jacobsen and M. Pellegrini (2012) Transcriptome and methylome interactions in rice hybrids. Proc. Natl. Acad. Sci. USA 109: 12040–12045.
- Cortijo, S., R. Wardenaar, M. Colomé-Tatché, A. Gilly, M. Etcheverry, K. Labadie, E. Caillieux, F. Hospital, J.M. Aury, P. Wincker et al. (2014) Mapping the epigenetic basis of complex traits. Science 343: 1145–1148.
- Crow, J.F. (1998) 90 years ago: The beginning of hybrid maize. Genetics 148: 923–928.
- Cui, J., C. You and X. Chen (2017) The evolution of microRNAs in plants. Curr. Opin. Plant Biol. 35: 61–67.
- Dapp, M., J. Reinders, A. Bédiée, C. Balsera, E. Bucher, G. Theiler, C. Granier and J. Paszkowski (2015) Heterosis and inbreeding depression of epigenetic Arabidopsis hybrids. Nat. Plants 1: 15092.
- Davenport, C.B. (1908) Degeneration, albinism and inbreeding. Science 28: 454–455.
- de la Rosa Santamaria, R., M.R. Shao, G. Wang, D.O. Nino-Liu, H. Kundariya, Y. Wamboldt, I. Dweikat and S.A. Mackenzie (2014) MSH1-induced non-genetic variation provides a source of phenotypic diversity in Sorghum bicolor. PLoS ONE 9: e108407.
- Duvick, D.N. (2001) Biotechnology in the 1930s: the development of hybrid maize. Nat. Rev. Genet. 2: 69–74.
- East, E.M. (1936) Heterosis. Genetics 21: 375–397.
- Frascaroli, E., M.A. Canè, P. Landi, G. Pea, L. Gianfranceschi, M. Villa, M. Morgante and M.E. Pè (2007) Classical genetic and quantitative trait loci analyses of heterosis in a maize hybrid between two elite inbred lines. Genetics 176: 625–644.
- Fujimoto, R., T. Sasaki, H. Inoue and T. Nishio (2008) Hypomethylation and transcriptional reactivation of retrotransposon-like sequences in ddm1 transgenic plants of Brassica rapa. Plant Mol. Biol. 66: 463–473.
- Fujimoto, R., J.M. Taylor, T. Sasaki, T. Kawanabe and E.S. Dennis (2011) Genome wide gene expression in artificially synthesized amphidiploids of Arabidopsis. Plant Mol. Biol. 77: 419–431.
- Fujimoto, R., J.M. Taylor, S. Shirasawa, W.J. Peacock and E.S. Dennis (2012a) Heterosis of Arabidopsis hybrids between C24 and Col is associated with increased photosynthesis capacity. Proc. Natl. Acad. Sci. USA 109: 7109–7114.
- Fujimoto, R., T. Sasaki, R. Ishikawa, K. Osabe, T. Kawanabe and E.S. Dennis (2012b) Molecular mechanisms of epigenetic variation in plants. Int. J. Mol. Sci. 13: 9900–9922.
- Giraud, H., C. Bauland, M. Falque, D. Madur, V. Combes, P. Jamin, C. Monteil, J. Laborde, C. Palaffre, A. Gaillard et al. (2017) Reciprocal genetics: Identifying QTL for general and specific combining abilities in hybrids between multiparental populations from two maize (Zea mays L.) heterotic groups. Genetics 207: 1167–1180.
- Girke, A., A. Schierholt and H.C. Becker (2012) Extending the rapeseed gene pool with resynthesized Brassica napus II: Heterosis. Theor. Appl. Genet. 124: 1017–1026.
- Greaves, I.K., M. Groszmann, H. Ying, J.M. Taylor, W.J. Peacock and E.S. Dennis (2012) Trans chromosomal methylation in Arabidopsis hybrids. Proc. Natl. Acad. Sci. USA 109: 3570–3575.
- Greaves, I.K., R. Gonzalez-Bayon, L. Wang, A. Zhu, P.C. Liu, M. Groszmann, W.J. Peacock and E.S. Dennis (2015) Epigenetic changes in hybrids. Plant Physiol. 168: 1197–1205.
- Groszmann, M., I.K. Greaves, Z.I. Albertyn, G.N. Scofield, W.J. Peacock and E.S. Dennis (2011a) Changes in 24-nt siRNA levels in Arabidopsis hybrids suggest an epigenetic contribution to hybrid vigor. Proc. Natl. Acad. Sci. USA 108: 2617–2622.
- Groszmann, M., I.K. Greaves, N. Albert, R. Fujimoto, C.A. Helliwell, E.S. Dennis and W.J. Peacock (2011b) Epigenetics in plants-vernalisation and hybrid vigour. Biochim. Biophys. Acta 1809: 427–437.
- Groszmann, M., I.K. Greaves, R. Fujimoto, W.J. Peacock and E.S. Dennis (2013) The role of epigenetics in hybrid vigour. Trends Genet. 29: 684–690.
- Groszmann, M., R. Gonzalez-Bayon, I.K. Greaves, L. Wang, A.K. Huen, W.J. Peacock and E.S. Dennis (2014) Intraspecific Arabidopsis hybrids show different patterns of heterosis despite the close relatedness of the parental genomes. Plant Physiol. 166: 265–280.
- Groszmann, M., R. Gonzalez-Bayon, R.L. Lyons, I.K. Greaves, K. Kazan, W.J. Peacock and E.S. Dennis (2015) Hormone-regulated defense and stress response networks contribute to heterosis in Arabidopsis F1 hybrids. Proc. Natl. Acad. Sci. USA 112: E6397–6406.
- Guo, Q., Q. Liu, N.A. Smith, G. Liang and M.B. Wang (2016) RNA silencing in plants: Mechanisms, technologies and applications in horticultural crops. Curr. Genomics 17: 476–489.
- Ha, M., J. Lu, L. Tian, V. Ramachandran, K.D. Kasschau, E.J. Chapman, J.C. Carrington, X. Chen, X.J. Wang and Z.J. Chen (2009) Small RNAs serve as a genetic buffer against genomic shock in Arabidopsis interspecific hybrids and allopolyploids. Proc. Natl. Acad. Sci. USA 106: 17835–17840.
- Haag, J.R., T.S. Ream, M. Marasco, C.D. Nicora, A.D. Norbeck, L. Pasa-Tolic and C.S. Pikaard (2012) In vitro transcription activities of Pol IV, Pol V, and RDR2 reveal coupling of Pol IV and RDR2 for dsRNA synthesis in plant RNA silencing. Mol. Cell 48: 811–818.
- Haag, J.R., B. Brower-Toland, E.K. Krieger, L. Sidorenko, C.D. Nicora, A.D. Norbeck, A. Irsigler, H. LaRue, J. Brzeski, K. McGinnis et al. (2014) Functional diversification of maize RNA Polymerase IV and V subtypes via alternative catalytic subunits. Cell Rep. 9: 378–390.
- He, G., X. Zhu, A.A. Elling, L. Chen, X. Wang, L. Guo, M. Liang, H. He, H. Zhang, F. Chen et al. (2010) Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell 22: 17–33.
- Hollister, J.D., L.M. Smith, Y.L. Guo, F. Ott, D. Weigel and B.S. Gaut (2011) Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc. Natl. Acad. Sci. USA 108: 2322–2327.
- Hu, X., H. Wang, X. Diao, Z. Liu, K. Li, Y. Wu, Q. Liang, H. Wang and C. Huang (2016) Transcriptome profiling and comparison of maize ear heterosis during the spikelet and floret differentiation stages. BMC Genomics 17: 959.
- Huang, X., S. Yang, J. Gong, Q. Zhao, Q. Feng, Q. Zhan, Y. Zhao, W. Li, B. Cheng, J. Xia et al. (2016) Genomic architecture of heterosis for yield traits in rice. Nature 537: 629–633.
- Jahnke, S., B. Sarholz, A. Thiemann, V. Kühr, J.F. Gutiérrez-Marcos, H.H. Geiger, H.P. Piepho and S. Scholten (2010) Heterosis in early seed development: a comparative study of F1 embryo and endosperm tissues 6 days after fertilization. Theor. Appl. Genet. 120: 389–400.
- Jiang, Y., R.H. Schmidt, Y. Zhao and J.C. Reif (2017) A quantitative genetic framework highlights the role of epistatic effects for grain-yield heterosis in bread wheat. Nat. Genet. 49: 1741–1746.
- Johannes, F., E. Porcher, F.K. Teixeira, V. Saliba-Colombani, M. Simon, N. Agier, A. Bulski, J. Albuisson, F. Heredia, P. Audigier et al. (2009) Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet. 5: e1000530.
- Jones, D.F. (1917) Dominance of linked factors as a means of accounting for heterosis. Proc. Natl. Acad. Sci. USA 3: 310–312.
- Kawamura, K., T. Kawanabe, M. Shimizu, K. Okazaki, M. Kaji, E.S. Dennis, K. Osabe and R. Fujimoto (2015) Genetic characterization of inbred lines of Chinese cabbage by DNA markers; towards the application of DNA markers to breeding of F1 hybrid cultivars. Data Brief 6: 229–237.
- Kawamura, K., T. Kawanabe, M. Shimizu, A.J. Nagano, N. Saeki, K. Okazaki, M. Kaji, E.S. Dennis, K. Osabe and R. Fujimoto (2016) Genetic distance of inbred lines of Chinese cabbage and its relationship to heterosis. Plant Gene 5: 1–7.
- Kawanabe, T., S. Ishikura, N. Miyaji, T. Sasaki, L.M. Wu, E. Itabashi, S. Takada, M. Shimizu, T. Takasaki-Yasuda, K. Osabe et al. (2016) Role of DNA methylation in hybrid vigor in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 113: E6704–E6711.
- Kempe, K., M. Rubtsova and M. Gils (2014) Split-gene system for hybrid wheat seed production. Proc. Natl. Acad. Sci. USA 111: 9097–9102.
- Kenan-Eichler, M., D. Leshkowitz, L. Tal, E. Noor, C. Melamed-Bessudo, M. Feldman and A.A. Levy (2011) Wheat hybridization and polyploidization results in deregulation of small RNAs. Genetics 188: 263–272.
- Ko, D.K., D. Rohozinski, Q. Song, S.H. Taylor, T.E. Juenger, F.G. Harmon and Z.J. Chen (2016) Temporal shift of circadian-mediated gene expression and carbon fixation contributes to biomass heterosis in maize hybrids. PLoS Genet. 12: e1006197.
- Korn, M., S. Peterek, H.P. Mock, A.G. Heyer and D.K. Hincha (2008) Heterosis in the freezing tolerance, and sugar and flavonoid contents of crosses between Arabidopsis thaliana accessions of widely varying freezing tolerance. Plant Cell Environ. 31: 813–827.
- Korn, M., T. Gärtner, A. Erban, J. Kopka, J. Selbig and D.K. Hincha (2010) Predicting Arabidopsis freezing tolerance and heterosis in freezing tolerance from metabolite composition. Mol. Plant 3: 224–235.
- Krieger, U., Z.B. Lippman and D. Zamir (2010) The flowering gene SINGLE FLOWER TRUSS drives heterosis for yield in tomato. Nat. Genet. 42: 459–463.
- Kurihara, Y., A. Matsui, M. Kawashima, E. Kaminuma, J. Ishida, T. Morosawa, Y. Mochizuki, N. Kobayashi, T. Toyoda, K. Shinozaki et al. (2008) Identification of the candidate genes regulated by RNA-directed DNA methylation in Arabidopsis. Biochem. Biophys. Res. Commun. 376: 553–557.
- Lauss, K., R. Wardenaar, M.H.A. van Hulten, V. Guryev, J.J.B. Keurentjes, M. Stam and F. Johannes (2016) Epigenetic divergence is sufficient to trigger heterosis in Arabidopsis thaliana. bioRxiv.
- Law, J.A., A.A. Vashisht, J.A. Wohlschlegel and S.E. Jacobsen (2011) SHH1, a homeodomain protein required for DNA methylation, as well as RDR2, RDM4, and chromatin remodeling factors, associate with RNA polymerase IV. PLoS Genet. 7: e1002195.
- Li, A., D. Liu, J. Wu, X. Zhao, M. Hao, S. Geng, J. Yan, X. Jiang, L. Zhang, J. Wu et al. (2014) mRNA and small RNA transcriptomes reveal insights into dynamic homoeolog regulation of allopolyploid heterosis in nascent hexaploid wheat. Plant Cell 26: 1878–1900.
- Li, D., Z. Huang, S. Song, Y. Xin, D. Mao, Q. Lv, M. Zhou, D. Tian, M. Tang, Q. Wu et al. (2016) Integrated analysis of phenome, genome, and transcriptome of hybrid rice uncovered multiple heterosis-related loci for yield increase. Proc. Natl. Acad. Sci. USA 113: E6026–E6035.
- Li, H., Q. Yang, N. Fan, M. Zhang, H. Zhai, Z. Ni and Y. Zhang (2017) Quantitative trait locus analysis of heterosis for plant height and ear height in an elite maize hybrid zhengdan 958 by design III. BMC Genet. 18: 36.
- Li, L., K. Lu, Z. Chen, T. Mu, Z. Hu and X. Li (2008) Dominance, overdominance and epistasis condition the heterosis in two heterotic rice hybrids. Genetics 180: 1725–1742.
- Li, Y., K. Varala, S.P. Moose and M.E. Hudson (2012) The inheritance pattern of 24 nt siRNA clusters in arabidopsis hybrids is influenced by proximity to transposable elements. PLoS ONE 7: e47043.
- Li, Z.K., L.J. Luo, H.W. Mei, D.L. Wang, Q.Y. Shu, R. Tabien, D.B. Zhong, C.S. Ying, J.W. Stansel, G.S. Khush et al. (2001) Overdominant epistatic loci are the primary genetic basis of inbreeding depression and heterosis in rice. I. Biomass and grain yield. Genetics 158: 1737–1753.
- Lippman, Z.B. and D. Zamir (2007) Heterosis: revisiting the magic. Trends Genet. 23: 60–66.
- Lu, H., J. Romero-Severson and R. Bernardo (2003) Genetic basis of heterosis explored by simple sequence repeat markers in a randommated maize population. Theor. Appl. Genet. 107: 494–502.
- Luo, L.J., Z.K. Li, H.W. Mei, Q.Y. Shu, R. Tabien, D.B. Zhong, C.S. Ying, J.W. Stansel, G.S. Khush and A.H. Paterson (2001) Overdominant epistatic loci are the primary genetic basis of inbreeding depression and heterosis in rice. II. Grain yield components. Genetics 158: 1755–1771.
- Meehan, L., K. Harkins, J. Chory and S. Rodermel (1996) Lhcb transcription is coordinated with cell size and chlorophyll accumulation. Plant Physiol. 112: 953–963.
- Meyer, R.C., H. Witucka-Wall, M. Becher, A. Blacha, A. Boudichevskaia, P. Dörmann, O. Fiehn, S. Friedel, M. von Korff, J. Lisec et al. (2012) Heterosis manifestation during early Arabidopsis seedling development is characterized by intermediate gene expression and enhanced metabolic activity in the hybrids. Plant J. 71: 669–683.
- Miedaner, T., A.W. Schulthess, M. Gowda, J.C. Reif and C.F. Longin (2017) High accuracy of predicting hybrid performance of Fusarium head blight resistance by mid-parent values in wheat. Theor. Appl. Genet. 130: 461–470.
- Miller, M., Q. Song, X. Shi, T.E. Juenger and Z.J. Chen (2015) Natural variation in timing of stress-responsive gene expression predicts heterosis in intraspecific hybrids of Arabidopsis. Nat. Commun. 6: 7453.
- Moll, R.H., J.H. Lonnquist, J. Vélez Fortuno and E.C. Johnson (1965) The relationship of heterosis and genetic divergence in maize. Genetics 52: 139–144.
- Ng, D.W., M. Miller, H.H. Yu, T.Y. Huang, E.D. Kim, J. Lu, Q. Xie, C.R. McClung and Z.J. Chen (2014) A role for CHH methylation in the parent-of-origin effect on altered circadian rhythms and biomass heterosis in Arabidopsis intraspecific hybrids. Plant Cell 26: 2430–2440.
- Ni, Z., E.D. Kim, M. Ha, E. Lackey, J. Liu, Y. Zhang, Q. Sun and Z.J. Chen (2009) Altered circadian rhythms regulate growth vigour in hybrids and allopolyploids. Nature 457: 327–331.
- Osabe, K., T. Kawanabe, T. Sasaki, R. Ishikawa, K. Okazaki, E.S. Dennis, T. Kazama and R. Fujimoto (2012a) Multiple mechanisms and challenges for the application of allopolyploidy in plants. Int. J. Mol. Sci. 13: 8696–8721.
- Osabe, K., T. Sasaki, R. Ishikawa and R. Fujimoto (2012b) The role of DNA methylation in plants. In: Tatiana, T.V. and G. Sablok (eds.) DNA Methylation: Principles, Mechanisms and Challenges. Nova Science Publishers, USA, pp. 35–66.
- Ossowski, S., K. Schneeberger, J.I. Lucas-Lledó, N. Warthmann, R.M. Clark, R.G. Shaw, D. Weigel and M. Lynch (2010) The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 327: 92–94.
- Paschold, A., Y. Jia, C. Marcon, S. Lund, N.B. Larson, C.T. Yeh, S. Ossowski, C. Lanz, D. Nettleton, P.S. Schnable et al. (2012) Complementation contributes to transcriptome complexity in maize (Zea mays L.) hybrids relative to their inbred parents. Genome Res. 22: 2445–2454.
- Powers, L. (1944) An expansion of Jones’s theory for the explanation of heterosis. Am. Nat. 78: 275–280.
- Pu, Z., M. Shimizu, Y. Zhang, T. Nagaoka, T. Hayashi, H. Hori, S. Matsumoto, R. Fujimoto and K. Okazaki (2012) Genetic mapping of a fusarium wilt resistance gene in Brassica oleracea. Mol. Breed. 30: 809–818.
- Pyke, K.A. and R.M. Leech (1991) Rapid image analysis screening procedure for identifying chloroplast number mutants in mesophyll cells of Arabidopsis thaliana (L.) Heynh. Plant Physiol. 96: 1193–1195.
- Reinders, J., B.B.H. Wulff, M. Mirouze, A. Marí-Ordóñez, M. Dapp, W. Rozhon, E. Bucher, G. Theiler and J. Paszkowski (2009) Compromised stability of DNA methylation and transposon immobilization in mosaic Arabidopsis epigenomes. Genes Dev. 23: 939–950.
- Richey, F.D. (1942) Mock-dominance and hybrid vigor. Science 96: 280–281.
- Rigal, M., C. Becker, T. Pélissier, R. Pogorelcnik, J. Devos, Y. Ikeda, D. Weigel and O. Mathieu (2016) Epigenome confrontation triggers immediate reprogramming of DNA methylation and transposon silencing in Arabidopsis thaliana F1 epihybrids. Proc. Natl. Acad. Sci. USA 113: E2083–2092.
- Rohde, P., D.K. Hincha and A.G. Heyer (2004) Heterosis in the freezing tolerance of crosses between two Arabidopsis thaliana accessions (Columbia-0 and C24) that show differences in non-acclimated and acclimated freezing tolerance. Plant J. 38: 790–799.
- Saeki, N., T. Kawanabe, H. Ying, M. Shimizu, M. Kojima, H. Abe, K. Okazaki, M. Kaji, J.M. Taylor, H. Sakakibara et al. (2016) Molecular and cellular characteristics of hybrid vigour in a commercial hybrid of Chinese cabbage. BMC Plant Biol. 16: 45.
- Sasaki, T., R. Fujimoto, S. Kishitani and T. Nishio (2011) Analysis of target sequences of DDM1s in Brassica rapa by MSAP. Plant Cell Rep. 30: 81–88.
- Schmitz, R.J., M.D. Schultz, M.G. Lewsey, R.C. O’Malley, M.A. Urich, O. Libiger, N.J. Schork and J.R. Ecker (2011) Transgenerational epigenetic instability is a source of novel methylation variants. Science 334: 369–373.
- Shen, H., H. He, J. Li, W. Chen, X. Wang, L. Guo, Z. Peng, G. He, S. Zhong, Y. Qi et al. (2012) Genome-wide analysis of DNA methylation and gene expression changes in two Arabidopsis ecotypes and their reciprocal hybrids. Plant Cell 24: 875–892.
- Shen, Y., S. Sun, S. Hua, E. Shen, C.Y. Ye, D. Cai, M.P. Timko, Q.H. Zhu and L. Fan (2017) Analysis of transcriptional and epigenetic changes in hybrid vigor of allopolyploid Brassica napus uncovers key roles for small RNAs. Plant J. 91: 874–893.
- Shimizu, M., R. Fujimoto, H. Ying, Z.J. Pu, Y. Ebe, T. Kawanabe, N. Saeki, J.M. Taylor, M. Kaji, E.S. Dennis et al. (2014) Identification of candidate genes for Fusarium yellows resistance in Chinese cabbage by differential expression analysis. Plant Mol. Biol. 85: 247–257.
- Shimizu, M., Z.J. Pu, T. Kawanabe, H. Kitashiba, S. Matsumoto, Y. Ebe, M. Sano, T. Funaki, E. Fukai, R. Fujimoto et al. (2015) Map-based cloning of a candidate gene conferring Fusarium yellows resistance in Brassica oleracea. Theor. Appl. Genet. 128: 119–130.
- Shull, G.H. (1908) The composition of a field of maize. J. Hered. 4: 296–301.
- Shull, G.H. (1948) What Is “Heterosis”? Genetics 33: 439–446.
- Song, G.S., H.L. Zhai, Y.G. Peng, L. Zhang, G. Wei, X.Y. Chen, Y.G. Xiao, L. Wang, Y.J. Chen, B. Wu et al. (2010) Comparative transcriptional profiling and preliminary study on heterosis mechanism of super-hybrid rice. Mol. Plant 3: 1012–1025.
- Springer, N.M. and R.M. Stupar (2007) Allelic variation and heterosis in maize: how do two halves make more than a whole? Genome Res. 17: 264–275.
- Springer, N.M. (2013) Epigenetics and crop improvement. Trends Genet. 29: 241–247.
- Stuber, C.W., S.E. Lincoln, D.W. Wolff, T. Helentjaris and E.S. Lander (1992) Identification of genetic factors contributing to heterosis in a hybrid from two elite maize inbred lines using molecular markers. Genetics 132: 823–839.
- Stupar, R.M., J.M. Gardiner, A.G. Oldre, W.J. Haun, V.L. Chandler and N.M. Springer (2008) Gene expression analyses in maize inbreds and hybrids with varying levels of heterosis. BMC Plant Biol. 8: 33.
- Swanson-Wagner, R.A., Y. Jia, R. DeCook, L.A. Borsuk, D. Nettleton and P.S. Schnable (2006) All possible modes of gene action are observed in a global comparison of gene expression in a maize F1 hybrid and its inbred parents. Proc. Natl. Acad. Sci. USA 103: 6805–6810.
- Tazoe, Y., T. Sazuka, M. Yamaguchi, C. Saito, M. Ikeuchi, K. Kanno, S. Kojima, K. Hirano, H. Kitano, S. Kasuga et al. (2016) Growth properties and biomass production in the hybrid C4 crop Sorghum bicolor. Plant Cell Physiol. 57: 944–952.
- Teixeira, F.K., F. Heredia, A. Sarazin, F. Roudier, M. Boccara, C. Ciaudo, C. Cruaud, J. Poulain, M. Berdasco, M.F. Fraga et al. (2009) A role for RNAi in the selective correction of DNA methylation defects. Science 323: 1600–1604.
- Tonosaki, K., K. Osabe, T. Kawanabe and R. Fujimoto (2016) The importance of reproductive barriers and the effect of allopolyploidization on crop breeding. Breed. Sci. 66: 333–349.
- Virdi, K.S., J.D. Laurie, Y.Z. Xu, J. Yu, M.R. Shao, R. Sanchez, H. Kundariya, D. Wang, J.J. Riethoven, Y. Wamboldt et al. (2015) Arabidopsis MSH1 mutation alters the epigenome and produces heritable changes in plant growth. Nat. Commun. 6: 6386.
- Vongs, A., T. Kakutani, R.A. Martienssen and E.J. Richards (1993) Arabidopsis thaliana DNA methylation mutants. Science 260: 1926–1928.
- Wang, L., I.K. Greaves, M. Groszmann, L.M. Wu, E.S. Dennis and W.J. Peacock (2015) Hybrid mimics and hybrid vigor in Arabidopsis. Proc. Natl. Acad. Sci. USA 112: E4959–E4967.
- Wang, L., L.M. Wu, I.K. Greaves, A. Zhu, E.S. Dennis and W.J. Peacock (2017) PIF4-controlled auxin pathway contributes to hybrid vigor in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 114: E3555–E3562.
- Wei, G., Y. Tao, G. Liu, C. Chen, R. Luo, H. Xia, Q. Gan, H. Zeng, Z. Lu, Y. Han et al. (2009) A transcriptomic analysis of superhybrid rice LYP9 and its parents. Proc. Natl. Acad. Sci. USA 106: 7695–7701.
- Wendte, J.M. and C.S. Pikaard (2017) The RNAs of RNA-directed DNA methylation. Biochim. Biophys. Acta 1860: 140–148.
- Williams, W. (1959) Heterosis and the genetics of complex characters. Nature 184: 527–530.
- Xiao, J., J. Li, L. Yuan and S.D. Tanksley (1995) Dominance is the major genetic basis of heterosis in rice as revealed by QTL analysis using molecular markers. Genetics 140: 745–754.
- Xu, Y.Z., M.P. Arrieta-Montiel, K.S. Virdi, W.B. de Paula, J.R. Widhalm, G.J. Basset, J.I. Davila, T.E. Elthon, C.G. Elowsky, S.J. Sato et al. (2011) MutS HOMOLOG1 is a nucleoid protein that alters mitochondrial and plastid properties and plant response to high light. Plant Cell 23: 3428–3441.
- Xu, Y.Z., R. de la Rosa Santamaria, K.S. Virdi, M.P. Arrieta-Montiel, F. Razvi, S. Li, G. Ren, B. Yu, D. Alexander, L. Guo et al. (2012) The chloroplast triggers developmental reprogramming when mutS HOMOLOG1 is suppressed in plants. Plant Physiol. 159: 710–720.
- Yang, L., B. Li, X.Y. Zheng, J. Li, M. Yang, X. Dong, G. He, C. An and X.W. Deng (2015a) Salicylic acid biosynthesis is enhanced and contributes to increased biotrophic pathogen resistance in Arabidopsis hybrids. Nat. Commun. 6: 7309.
- Yang, M., X. Wang, D. Ren, H. Huang, M. Xu, G. He and X.W. Deng (2017) Genomic architecture of biomass heterosis in Arabidopsis. Proc. Natl. Acad. Sci. USA 114: 8101–8106.
- Yang, X., H. Kundariya, Y.Z. Xu, A. Sandhu, J. Yu, S.F. Hutton, M. Zhang and S.A. Mackenzie (2015b) MutS HOMOLOG1-derived epigenetic breeding potential in tomato. Plant Physiol. 168: 222–232.
- Zemach, A., M.Y. Kim, P.H. Hsieh, D. Coleman-Derr, L. Eshed-Williams, K. Thao, S.L. Harmer and D. Zilberman (2013) The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 153: 193–205.
- Zhai, R., Y. Feng, H. Wang, X. Zhan, X. Shen, W. Wu, Y. Zhang, D. Chen, G. Dai, Z. Yang et al. (2013) Transcriptome analysis of rice root heterosis by RNA-Seq. BMC Genomics 14: 19.
- Zhang, Q., D. Wang, Z. Lang, L. He, L. Yang, L. Zeng, Y. Li, C. Zhao, H. Huang, H. Zhang et al. (2016a) Methylation interactions in Arabidopsis hybrids require RNA-directed DNA methylation and are influenced by genetic variation. Proc. Natl. Acad. Sci. USA 113: E4248–E4256.
- Zhang, Q., Y. Li, T. Xu, A.K. Srivastava, D. Wang, L. Zeng, L. Yang, L. He, H. Zhang, Z. Zheng et al. (2016b) The chromatin remodeler DDM1 promotes hybrid vigor by regulating salicylic acid metabolism. Cell Discov. 2: 16027.
- Zhao, Y., M.F. Mette and J.C. Reif (2015) Genomic selection in hybrid breeding. Plant Breed. 134: 1–10.
- Zhen, G., P. Qin, K.Y. Liu, D.Y. Nie, Y.Z. Yang, X.W. Deng and H. He (2017) Genome-wide dissection of heterosis for yield traits in two-line hybrid rice populations. Sci. Rep. 7: 7635.
- Zhu, A., I.K. Greaves, P.C. Liu, L. Wu, E.S. Dennis and W.J. Peacock (2016) Early changes of gene activity in developing seedlings of Arabidopsis hybrids relative to parents may contribute to hybrid vigour. Plant J. 88: 597–607.
