Abstract
Precision medicine is an approach to developing drugs that focuses on employing biomarkers to stratify patients in clinical trials with the goal of improving efficacy and/or safety outcomes, ultimately increasing the odds of clinical success and drug approval. Precision medicine is an important tool for toxicologists to utilize, because its principles can be used to decide whether to pursue a drug target, to understand interindividual differences in response to drugs in both nonclinical and clinical settings, to aid in selecting doses that optimize efficacy or reduce adverse events, and to facilitate understanding of a drug’s mode-of-action. Nonclinical models such as the mouse and non-human primate can be used to understand genetic variation and its potential translation to humans, and are available for toxicologists to employ in advance of drugs moving into clinical development. Understanding interindividual differences in response to drugs and how these differences can influence the drug’s risk-benefit profile and lead to the identification of biomarkers that enhance patient efficacy and safety is of critical importance for toxicologists today, and in the future, as the fields of pharmacogenomics and genetics continue to advance.
Abbreviations
6-MP = 6-mercaptopurine
ADME = Absorption, distribution, metabolism, and excretion
ALT = Alanine aminotransferase
CETPi = Cholesterol ester transfer protein inhibitor
CNV = Copy number variant
CYP = Cytochrome P450
GOF = Gain of function
DILI = Drug-induced liver injury
GWAS = Genome-wide association studies
HDL = High-density lipoprotein
HIV = Human immunodeficiency virus
INDELS = Insertion and deletion
LOF = Loss of function
MMHP = Mouse model of the human population
MOA = Mode-of-action
MPS = Microphysiological system
MDR = Multidrug resistence
MRP = Multidrug resistance-associated protein
OATP = Organic anion transporting polypeptide
NUD15 = Nucleoside diphosphatase-15
NSCLC = Non-small cell lung carcinoma
R&D = Research and Development
SLCO = Solute carrier organic anion
SNPs = Single nucleotide polymorphisms
TPMT = Thiopurine S-methyltransferase
VLDL = Very-low density lipoprotein
INTRODUCTION
Precision medicine is an approach to developing drugs that uses biomarkers to stratify patients for treatment, resulting in an enhanced efficacy and/or safety profile for the drug and improved clinical trial outcome (Dolsten and Søgaard, 2012). Commonly used terms in precision medicine are listed in Table 1 and several excellent reviews that explain this field have been published (Court, 2007; Pletcher et al., 2010; Sissung et al., 2012; Tzvetkov and von Ahsen, 2012). The ultimate goal of precision medicine is to increase the chances of having a successful clinical trial and obtaining drug approval. In contrast, personalized medicine is a vision of medical practice where the unique aspects of a patient, including his or her genetic make-up, are considered in the optimization of their treatment (Dolsten and Søgaard, 2012). In precision medicine, the focus is on unique traits in the population while in personalized medicine the focus is on unique traits of the individual. The treatments and companion diagnostics developed through precision medicine facilitate the practice of personalized medicine (Table 2). Precision medicine is an important tool for toxicologists, because it can improve safety assessments, reduce pharmaceutical attrition and development time, and facilitate understanding of mode-of-action (MOA) of a drug. Therefore, toxicologists should be knowledgeable of and trained in precision medicine principles. This review will provide examples of applying precision medicine principles in drug development to improve R&D productivity from target selection through the Phase 4 postmarket period.
Table 1. Definitions of commonly used terms in precision medicine.
| Term |
Definition |
| Allelic Variants |
are differences between alleles, detectable in any assay. DNA variants do not always confer an external phenotype on the animal and are referred to as polymorphisms if their frequency is greater than 1% in the population. |
| Codon |
contains three bases in a DNA or RNA sequence that specify a single amino acid. |
| Exon |
is the region of a gene that contains the code for producing the gene’s protein. A gene’s exons are separated by long regions of non-coding DNA called introns. |
| Extensive Metabolizer |
is an individual who has at least one full functioning allele of a gene coding for a drug-metabolizing enzyme. |
| Genetic Variation |
is the variation in the DNA sequence between members of a population or species. Variation may be a single base pair to changes in whole genetic segments as a result of rearrangement, duplication, or deletion. |
| Genome |
is an organism’s genetic material. The human genome is the DNA contained in the chromosomes, totaling about 3 billion base pairs. |
| Genotype |
is the description of the genetic composition of the animal, usually in terms of particular alleles at a particular loci. |
| Genome-wide Association Study (GWAS) |
is a study that identifies markers across genomes to find genetic variation associated with a disease or condition. |
| Haplotype |
is a set of variants of closely-linked genetic loci that tend to be inherited together. |
| Heterozygous |
possesses two different copies of a particular gene |
| Homozygous |
possesses two identical copies of a particular gene (e.g., wild type, recessive) |
| Intermediate metabolizer |
is an individual with genotype coding for no enzyme function from one allele and reduced activity from the other allele |
| Intron |
is a noncoding sequence of DNA that initially is copied into RNA but is cut out of the final RNA transcript. |
| Pharmacogenetics |
is the study of drug interactions with a relatively restricted number of genes. |
| Pharmacogenomics |
is the study of drug interactions across the entire genome. |
| Phenotype |
is the result of interaction between genotype and the environment and can be determined by any assay. |
| Poor metabolizer |
is an individual with genotype coding for no or very little enzyme function from both alleles. |
| Single nucleotide polymorphisms (SNPs) |
are DNA sequence variations caused by single base changes at a given position in a genome. These substitution mutations occur at an average frequency of 1 SNP for every 1000 base pairs of DNA sequence. |
| Ultra Rapid Metabolizer |
is an individual who has excess enzymatic activity due to multiple copies of functional alleles from gene duplication; commonly seen with CYP2D6. |
(Court, 2007; Pletcher et al., 2010)
Table 2. Precision medicine biomarkers for drugs
(a).
| Biomarker |
Drug(s) |
| ALK(c) |
Alectinib, Brentuximab, Brigatinib, Ceritinib, Crizotinib |
| BCHE(b) |
Succinylcholine |
| BCR-ABL1(c) |
Blinatumomab, Bosutinib, Busulfan, Dasatinib, Imatinib, Inotuzumab, Nilotinib, Omacetaxine, Ponatinib |
| BRAF(c) |
Cobimetinib, Dabrafenib, Nivolumab, Pembrolizumab, Trametinib, Vemurafenib |
| BRCA(c) |
Niraparib, Olaparib, Rucaparib |
| CASR(b) |
Parathyroid hormone |
| CFTR(c) |
Ivacaftor, Lumcaftor |
| Chromosome 5q(c) |
Lenalidomide |
| Chromosome 11q(c) |
Ibrutinib |
| Chromosome 17p(c) |
Ibrutinib, Venetoclax |
| CYB5R(b) |
Metoclopramide, Primaquine, Rasburicase |
| CYP2B6(d) |
Efavirenz |
| CYP2C9(d) |
Celecoxib, Dronabinol, Flurbiprofen, Lesinurad, Phenytoin, Piroxicam, Warfarin |
| CYP2C19(d) |
Citalopram, Clobazam, Clopidogrel(e), Carisoprodol, Dexiansoprazole, Diazepam, Doxepin, Escitalopram, Omeprazole, Pantoprazole, Phenytoin, Voriconazole |
| CYP2D6(d) |
Amitriptyline, Aripiprazole, Atomoxetine, Brexpiprazole, Carvedilol, Cevimeline, Clomipramine, Clozapine, Codeine, Desipramine, Deutetrabenazine, Dextromethorphan, Doxepin, Duloxetine, Eliglustat, Fluoxetine, Iloperidone, Metoprolol, Nortriptyline, Paroxetine, Perphenazine, Pimozide(e), Propafenone, Propanolol, Protriptyline, Quinidine, Tetrabenazine, Tolterodine, Tramadol, Valbenazine, Vortioxetine |
| DPYD (DPD)(b) |
Capecitabine, Fluorouracil |
| DMD(c) |
Eteplirsen |
| EGFR(c) |
Afatinib, Cetuximab, Erlotinib, Gefinitib, Osimertinib, Panitumumab |
| ERRB2 (Her2)(c) |
Abemaciclib, Everolimus, Lapatinib, Neratinib, Palbociclib, Pertuzumab, Ribociclib, Trastuzumab |
| ESR (Estrogen receptor)(c) |
Abemaciclib, Anastrozole, Everolimus, Exemestane, Fulvestrant, Leutrozole, Palbociclib, Pertuzumab, Ribociclib, Tamoxifen, Trastuzumab |
| F5(c) |
Eltrombopag |
| FIP1L1-PDGFRα(c) |
Imatinib |
| FLT3(c) |
Midostaurin |
| GALNS(c) |
Elosulfase |
| G6PD(b) |
Ascorbic acid, Chloroquine, Chlorpropamide, Dabrafenib, Dapsone, Glimepiride, Glipizide, Glyburide, Metoclopramide, Mafenide, Methylene blue, Nalidixic acid, Nitrofurantoin, Pegloticase, Primaquine, Quinine sulfate, Rasburicase, Sodium Nitrite, Sulfasalazine, Trametinib |
| HLAs(b) |
Abacavir, Carbamazepine, Lapatinib, Oxcarbazepine, Pazopanib, Phenytoin |
| HPRT(b) |
Mycophenolic acid |
| IDH2(c) |
Enasidenib |
| IFNL3 (IL28B)(c) |
Boceprevir, Daclatasvir, Elbasvir, Grazoprevir, Ledipasvir, Ombitasvir, Peginterferon-alfa-2b, Simeprevir, Sofosbuvir, Telaprevir |
| IL2RA (CD25)(c) |
Denileukin, Diftitox |
| Kit(c) |
Imatinib, Midostaurin |
| Microsatellite instability(c) |
Nivolumab, Pembrolizumab |
| MYCN(c) |
Dinutuximab |
| MS4A1 (CD20) (c) |
Obinutuzumab, Rituximb |
| NAGS(c) |
Carglumic acid |
| NAT-1,2(d) |
Hydralazine, Isoniazid, Sulfamethoxazole, Sulfasalazine |
| NPM1(c) |
Midostaurin |
| PDGFR(c) |
Imatinib, Olaratumab |
| PD-L1 (CD274) (c) |
Atezolizumab, Avelomab, Durvalumab, Nivolumab, Pembrolizumab |
| PML/RARα(c) |
Arsenic trioxide, Tretinoin |
| POLG(b) |
Valproic Acid |
| PROC(b) |
Warfarin |
| PROS1(b) |
Warfarin |
| RAS(c) |
Cetuximab, Dabrafenib, Panitumumab, Trametinib, Vemurafenib |
| RET(c) |
Carbozantinib |
| ROS1(c) |
Crizotinib |
| SERPINC1(c) |
Eltrombopag |
| TPMT(b) |
Azathioprine, Cisplatin, Mercaptopurine, Thioguanine |
| TPP1(c) |
Cerliponase alfa |
| UGT1A1(d) |
Belinostat, Irinotecan, Nilotinib, Pazopanib |
| Urea Cycle Disorders(b) |
Valproic acid |
| VKORC1(d) |
Warfarin |
(a)FDA Table of Pharmacogenomic Biomarkers - December 2017 (https://www.fda.gov/Drugs/ScienceResearch/ucm572698.htm)
(b)Safety; (c)Efficacy; (d)ADME - Safety; (e)ADME - Efficacy
THE IMPORTANCE OF PRECISION MEDICINE
For the last several decades, the pharmaceutical industry has been experiencing a crisis in Research and Development (R&D) productivity. To develop a single drug requires on average 11 Phase 1 starts, development times of 9 years for small molecules and 10 years for biologics, and in excess of one billion US dollars (CMR Pharmaceutical International R&D Factbook, 2016). In a review of clinical trials from 2013-2015, lack of efficacy accounted for 48 and 55% of failure while safety issues accounted for 25 and 14% of failure in Phase 2 and 3 clinical trials, respectively (Harrison, 2016). Phase 2 clinical trials consume greater than 55% of R&D budgets. The efficacy attrition highlights why precision medicine and other tools such as microphysiological systems (MPS) (Ewart et al., 2018; Wang et al., 2017) and phenotypic screens (Moffat et al., 2017; Vincent et al., 2015) are needed.
Precision medicine is also needed, because a high percentage of patients using a particular drug class may derive no benefit from the drug (Spear et al., 2001). The percentage of patients where drug treatment is ineffective ranges from 38% for SSRI anti-depressants to 75% for oncology therapeutics (Spear et al., 2001). From an efficacy perspective, precision medicine has the opportunity to characterize complex diseases and identify targets to address unmet medical need, thereby increasing the percentage of patient responders. From a safety perspective, precision medicine has the opportunity to identify patient non-responders that experience no benefit from the drug but are still at risk for drug side effects, as well as identify early safety signals that can lead to an unacceptable risk/benefit profile.
Emerging science and technology to implement precision medicine has dramatically improved and is being used to facilitate targeting the right medicines to the right patient and thereby increasing the proportion of patient responders. The Human Genome Project cost one billion dollars and took nearly thirteen years (1990-2003) to sequence the human genomes of 12 subjects (Court, 2007; Pletcher et al., 2010; Varmus, 2010); the current cost of sequencing the human genome is $1100 ($500 for whole exome sequencing) and takes less than a month from submission of samples. It is these advancements that are allowing the practice of precision medicine to become a reality.
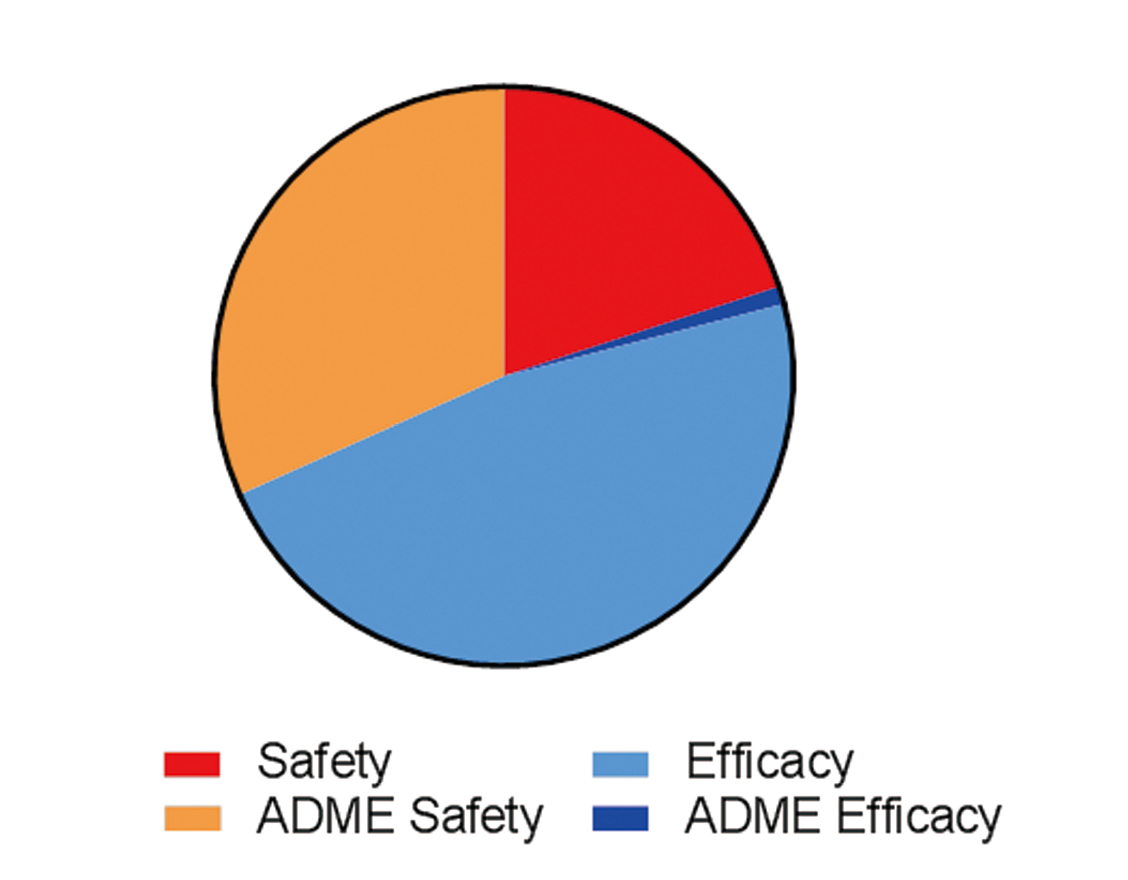
An analysis of the FDA Table of Pharmacogenomic Biomarkers in Drug Labels (https://www.fda.gov/Drugs/ScienceResearch/ucm572698.htm) was conducted (Table 2, Fig. 1). These biomarkers are being used to improve the safety or efficacy of drugs and four general categories were identified (Table 2): Safety, Efficacy, ADME Safety, and ADME Efficacy. Of the 205 biomarker/drug pairs, there are 51 precision medicine biomarkers currently in drug labels as of December 2017 (Table 2). Of these biomarker/drug pairs, 52% were used to exclude patients or adjust the therapeutic dose based on safety while 48% were used to select patients for efficacy (Fig. 1). Safety biomarkers represented 20% of the drugs while ADME Safety biomarkers, defined as those drugs whose dosage was adjusted to reduce adverse events, represented 32% of drugs (Fig. 1). Efficacy biomarkers were used to identify patients who would benefit from drugs and represented 47% of drugs while ADME Efficacy, defined as those drugs dependent on metabolism for converting a pro-drug into an active drug, represented 1% of the drugs (Fig. 1). ADME-related pharmacogenomics biomarkers represented 33% of the biomarker/drug pairs which reflect the large knowledge-base that exists for drug metabolizing enzymes (Phase I, II) and transporters (Phase III) (Grossman, 2009; Sissung et al., 2012). Some drugs have multiple biomarkers either for ADME (e.g., phenytoin - CYP2C9, CYP2C19) or efficacy (e.g., nivolumab – PD-L1, microsatellite instability). Hence, the evaluation of the genetic variants in a drug target is increasingly being applied to improve the safety or efficacy of a drug. To continue this momentum, DNA collection in clinical trials and exploratory biomarker wording added to informed consent documents needs to be routinely integrated into all clinical trials so that when interindividual differences in response are detected during clinical trials they can be studied for identification of potential pharmacogenomic biomarkers.
Classic examples of how precision medicine can positively impact drug development are highlighted by the drugs abacavir and crizotinib. Abacavir is a nucleoside reverse transcriptase inhibitor (NRTI) used to treat human immunodeficiency virus (HIV). Abacavir produced allergic hypersensitivity reactions that caused mortality in some patients. It was demonstrated that allelic carriers for HLA-B*5701 identified 95% of susceptible patients who were then excluded from using the drug or monitored more closely based on the results of the pharmacogenetic test for this allele (Hughes et al., 2008). For crizotinib, precision medicine was instrumental in speeding up development time. In 2007, the presence of the EML4-ALK fusion gene in non-small cell lung cancer (NSCLC) was first reported (Soda et al., 2007). This discovery led to clinical trials with the ALK inhibitor, crizotinib. In patients identified to have this mutation, tumors were not detectable upon X-ray after 2 cycles of crizotinib treatment (Kwak et al., 2009). Crizotinib was approved by the FDA in 2012, 5 years after the first description of the EML-4-ALK fusion gene in NSCLC. This timing is in contrast to the average development time of 9 years for a small molecule drug (CMR Pharmaceutical International R&D Factbook, 2016). In both of these examples, the application of precision medicine principles significantly increased patient benefit and drug success.
BASICS OF GENETICS
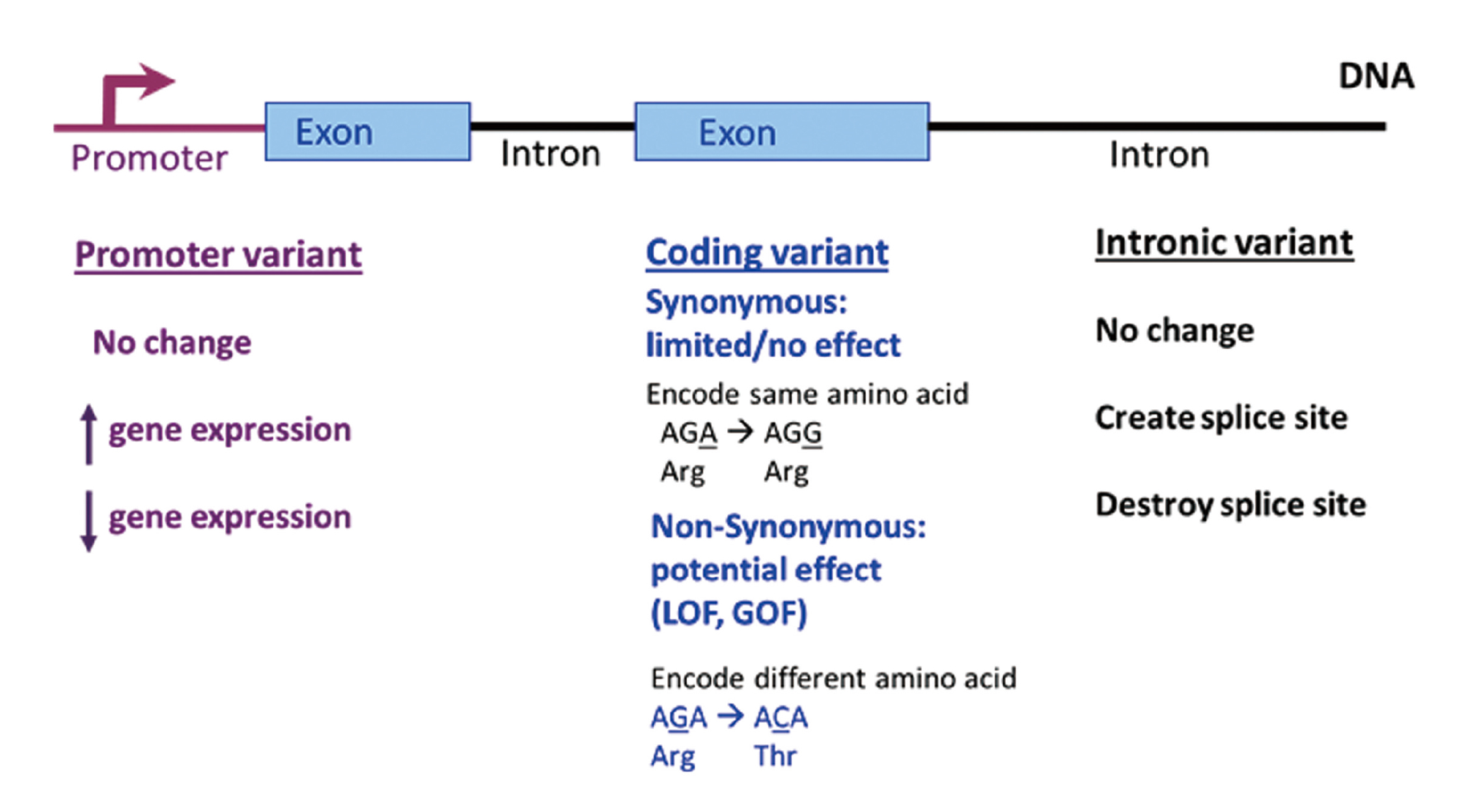
Genetics aims to identify and quantify the relationship between genotypes and phenotypes and several excellent overviews have been written that provide examples of its application to prescribing drugs (Court, 2007; Pletcher et al., 2010; Sissung et al., 2012; Tzvetkov and von Ahsen, 2012). The human genome contains 23 chromosome pairs of 3.1 billion base pairs and 23,500 protein coding genes. The protein coding genes represent only 1% of the genome. There are many types of genetic variations that contribute to individual variability in response to drugs, but single nucleotide polymorphisms (SNPs) are the most common. There are 15 million common SNPs expressed with an allelic frequency of greater than 5% and these are the primary focus of precision medicine-based biomarkers (Court, 2007). There is a much larger number of rare SNPs whose allelic frequency is less than 5%. The functional impact of a variant on gene function can be three-fold: gain of function (GOF), loss of function (LOF), or no change in function (Fig. 2). Promoter variants can decrease function (e.g., poor metabolizer variants), increase function (e.g., extensive or ultra-rapid metabolizer), or have no effect on gene expression and subsequent protein function (Mele and Goldschmidt, 2014; Turner, 2013). An example of promoter variant is a polymorphism that eliminates the ability of γ-interferon to regulate CYP2E1 activity by blocking its ability to bind to the response element within the 5’-regulatory region of CYP2E (Qiu et al., 2004). SNP coding variants can be either synonymous or non-synonymous. A synonymous coding variant has limited or no effect on protein function as the nucleotide change encodes for the same amino acid. A non-synonymous coding variant can result in GOF or LOF as the nucleotide change encodes a different amino acid. Intronic variants can create or destroy a splice site resulting in effects on the translated protein or have no effect. An example of this type of SNP is CYP3A5*3, which incorporates a portion of intron 3 resulting in low expression of CYP3A5 due to aberrant mRNA transcripts (Hustert et al., 2001; Kuehl et al., 2001). Two of the most common non-SNP allelic variations are insertion/deletion polymorphisms (commonly referred to as Indels) or copy number variations (CNV). Indels can produce a truncated protein when they cause a frame shift to produce a stop codon. The most common example of an indel is a microsatellite polymorphism that causes Gilberts’s syndrome (e.g., UGT1A1*28 polymorphism) (Nagar and Blanchard, 2006). CNVs in the CYP2D6 gene can result in a gene deletion producing a poor metabolizer phenotype (e.g., CYP2D6*5) or a gene duplication producing an ultra-rapid metabolizer phenotype by having 13 copies of CYP2D6 (Dalén et al., 1998; Ingelman-Sundberg et al., 1999).
Precision medicine has utilized two strategies to identify the genetic basis for individual variation in response to a drug: candidate gene association and genome-wide association studies (GWAS) (Mas and Lafuente, 2009). These studies are either exploratory or confirmatory in nature, independent of the design strategy. Prior to 2006, candidate gene studies were the principle tool used to discover variants and were selected based on a hypothesis of drug metabolism or mechanism. They focused on determining whether phenotypic variability between the case (e.g., nonresponder) and the control (e.g., responder) groups could be attributed to a gene variant. Candidate gene studies suffered from many false positive associations and well-powered studies required sample sizes in the thousands to more accurately determine an association coupled with functional studies to confirm the biologic association in order to correctly identify a phenotype/genotype association. It is now known that multiple polymorphisms exist in genes and characterizing the functionality of multiple polymorphisms is not possible using a candidate gene strategy. After 2006, GWAS became the norm, as they are an unbiased approach for assessing millions of variants across the entire human genome and their association with a defined phenotype. This strategy combines knowledge about linkage disequilibrium relationships and the way in which the genome is structured into haplotype blocks. This approach aims to identify the association between genetic variants and a given phenotype by detecting marker SNPs and analyzing differences between case and control groups. These data are typically visualized in Manhattan plots, and p values less than 10-8 are used as thresholds for identifying variants. GWAS also require multiple independent studies for confirmation and/or testing the biologic association through in vitro experiments to confirm the phenotype/genotype association.
NONCLINICAL APPROACHES TO ASSESS GENETIC DIVERSITY
Several preclinical models have been developed to assess the effects of genetic diversity and its translation to the human population. Mouse models include diversity panels such as the mouse model of the human population (MMHP) (Harrill et al., 2009), and collaborative cross (CC), and diversity outbred (DO) populations (Bogue et al., 2015; Morgan et al., 2015; Mosedale, 2018). The MMHP, a panel consisting of 36 different inbred mouse strains, was the first application of a mouse diversity panel in toxicology, with the objective to map polymorphisms that infer susceptility to acetaminophen-induced liver injury (Harrill et al., 2009). In this study, the CD44 gene was identified from haplotype-associated mapping conducted using ALT levels in the MMHP strains following treatment with a toxic dose of acetaminophen. CD44 was then verified to be a susceptibility factor for acetaminophen toxicity in humans using cohorts of subjects administered the maximum recommended therapeutic dose of acetaminophen (Court et al., 2014). The MMHP has also proven useful in identifying potential MOA for toxicants. Church and colleagues (Church et al., 2014) took a systems biology approach with the MMHP and identified that isoniazid-induced microvesicular steatosis is associated with mechanisms of oxidative stress, reduced export of lipids, and altered lipid packaging, which impart susceptibility for developing steatosis. Further, a mouse diversity panel was used to determine a potential mechanism for phospholipidosis induced by an antibiotic drug candidate (Mosedale et al., 2014).
While the MMHP studies clearly illustrated the value of using mouse diversity panels in toxicology, there are limitations in the genetic diversity that can be covered by the mouse panels. The CC initiative was a way to address this issue, with the goal of maximizing genetic diversity in purpose-bred mouse strains derived from independent breeding funnels originating with 8 inbred founder strains (A/J, C57BL6/J, 129S1/SvImJ, NOD/ShiLtJ, NZO/HlLtJ, CAST/EiJ, PWK/PhJ and WSB/EiJ) covering approximately 90% of known genetic diversity in Mus musculus (Threadgill and Churchill, 2012). Recent studies using the CC mice have highlighted their utility in identifying toxicological mechanisms. Hartman and colleagues (Hartman et al., 2017) used CC mouse strains to demonstrate that increased mitochondrial CYP 2E1 activity following exposure to butadiene resulted in decreased mitochondrial respiratory complex activity. Another study used the CC mice to evaluate tolvaptan-induced liver injury and identified associated susceptibility genes in bile acid homeostasis pathways (Mosedale et al., 2017). Tolvaptan was a promising drug candidate for the treatment of autosomal dominant polycystic kidney disease; however during clinical trials, DILI was observed in humans but not in species used in the preclinical safety assessment (Mosedale et al., 2017). The CC model replicated the liver injury in 3/45 strains and these strains identified polymorphisms in bile acid homeostasis as most associated with the liver injury, thereby providing potential biomarkers to manage DILI risk in these patients (Mosedale et al., 2017). In both of these examples, it was the natural variation of the studied phenotypes in the CC mice across an identical genetic background that was key to the findings being identified.
The DO mouse population was derived from some of the early breeding generations in the CC initiative, and is a tool that can be used in concert with the CC (Threadgill and Churchill, 2012). They are an outbred stock derived from the same eight inbred founder strains selected for the CC that accumulate recombination events with each outbreeding generation. The DO mice are maintained outbred through random mating which allows for very high-precision genetic mapping (Bogue et al., 2015). Because these mice are genetically unique, each mouse’s genome must be sequenced to enable QTL (quantitative trait locus) analysis, unlike the inbred mice utilized in diversity panels or the CC mouse strains. While the genetic polymorphisms in mice may not be identical to those in humans, one can expect to identify genes that provide mechanistic insight into the etiology of drug responses and toxicities that would not necessarily be identified in a single inbred model system. The DO mouse model has been used to investigate the DILI mechanisms observed in humans consuming green tea extract (Church et al., 2015). The likely causative agent, polyphenol epigallocatechin gallate (EGCG), was shown to to produce severe hepatotoxicity in 16% of the mice (43/272), a response rate similar to that observed in humans and providing the opportunity to identify potential causative polymorphisms.
The common advantage of all of these mouse models is the ability to recapitulate genetic variation observed in humans, and as many of the safety findings that limit drug use in the clinic are unique to small numbers of individuals, these are tools that are well positioned to advance the field of toxicology and enable precision medicine. The key limitations of these three mouse models are the large number of mice required to conduct these analyses and the limited endpoints that can be interogated. To address these limitations, in vitro platforms for these models are being developed using 3D models (Mosedale, 2018).
There is growing body of literature where whole exome sequencing data from non-human primates (e.g., cynomolgus monkey) can be used to select animals for immune oncology studies as well as to understand interindividual variability (Ebeling et al., 2011; Wu and Adkins, 2012; Yan et al., 2011). A recent example illustrates how genetic data can help understand interindividual variability in monkeys and how these data may be applied to protecting humans (Wu et al., 2017). Following treatment with metabotropic glutamate receptor 5 (mGluR5) negative allosteric modulators, clinical skin lesions were observed in Mauritius cynomolgus monkeys (Palanisamy et al., 2015). Similar skin lesions were also observed during preclinical development of a second drug candidate, where 9 out of 62 cynomolgus monkeys exhibited clinical and histopathologic skin effects that were consistent with characteristics of drug-induced skin reactions observed in humans (Wu et al., 2017). Microsatellite analysis identified 7 haplotypes and genetic association analysis identified alleles from the M3 haplotype of the MHC IB region (B*011:01, B*075:01, B*079:01, B*070:02, B*098:05, and B*165:01) to be significantly associated (χ2 test for trend, P < 0.05) with the occurrence of drug-induced hypersensitivity reactions in these monkeys. Sequence similarity from alignment of alleles in the M3 haplotype B region and HLA alleles associated with drug-induced hypersensitivity reactions in humans was 86 to 93% (Wu et al., 2017). These data demonstrate that MHC alleles in cynomolgus monkeys are associated with drug-induced hypersensitivity reactions, similar to HLA alleles in humans. Because of the high degree of sequence similarity between these alleles in the monkey M3 haplotype and human HLA alleles associated with drug-induced hypersensitivity reactions, identifying the specific alleles in the monkey may help in the identification of HLA alleles that may impart risk for patients administered these drugs. Therefore, understanding the genetic variations involved in the drug-induced adverse reactions in cynomolgus monkeys may predict human populations with susceptibility to drug-induced adverse events in clinic, thus decreasing drug development attrition and achieving the vision of precision medicine.
APPLYING PRECISION MEDICINE IN DRUG DEVELOPMENT
Target selection
Nelson and colleagues (Nelson et al., 2015) assessed whether genetic information influences drug survival rates using the Informa Pharmaprojects database. Analysis of 705 indications and a total of 19,085 target indication pairs demonstrated that the proportion of drug mechanisms with direct genetic support increases from 2% at the preclinical stage to 8.2% among mechanisms for approved drugs. Importantly, there was a 2-fold greater probability of success for a drug in Phase 1 to achieve regulatory approval when the target had genetic information that supported confidence in rationale. This was the first quantitative analysis to substantiate that genetics can reduce drug attrition. Therefore, incorporating genetic data into the target selection process can increase the probability of regulatory success.
Table 3 lists genetic databases that can be used to assess the efficacy and safety of targets. These databases can be used to assess safety of the target using a pharmacology analogy where LOF mutations are analogous to a drug being an antagonist to the target while GOF mutations are analogous to a drug being an agonist. In addition, known biological consequences from human LOF/GOF mutations can be used for comparison with mouse and rat knockout/knockin models of the pharmacologic target. If the animal model produces a similar phenotype to the human LOF/GOF mutation, there is increased confidence in using the animal model in early drug development.
Table 3. Human genetic databases used in assessing a target.
| Human Genetic Databases and Hyperlinks |
| Human Genetic Mutation Databases (HGMD) http://www.hgmd.cf.ac.uk/ac/index.php |
| Exome Variant Server (EVS) http://evs.gs.washington.edu/EVS/ |
| NHGRI GWAS catalogue http://www.genome.gov/26525384 |
| OMIM http://www.ncbi.nlm.nih.gov/omim |
| HuGE navigator http://hugenavigator.net/HuGENavigator/home.do |
| Disease Genetic Association Database (GAD) http://geneticassociationdb.nih.gov/cgi-bin/simple.cgi?table |
| Phenotype-Genotype Integrator http://www.ncbi.nlm.nih.gov/gap/PheGenI |
Discovery toxicology screening
Drug-induced liver injury (DILI) in patients has been associated with allelic variants in Phase I (e.g., CYP2E1), Phase II (e.g., NAT2, and GST) and Phase III (e.g., BSEP) metabolism (An et al., 2012; Lang et al., 2007; Rana et al., 2014; Teixeira et al., 2011). DILI is often multifactorial such as bosenten-induced DILI where allelic variants in Phase I (i.e., CYP2C9) and III (i.e., BSEP) metabolism appear to be causative (Fattinger et al., 2001; Markova et al., 2013). These examples suggest that understanding individual genetic variations involved with DILI could help the pharmaceutical industry understand at risk patient populations.
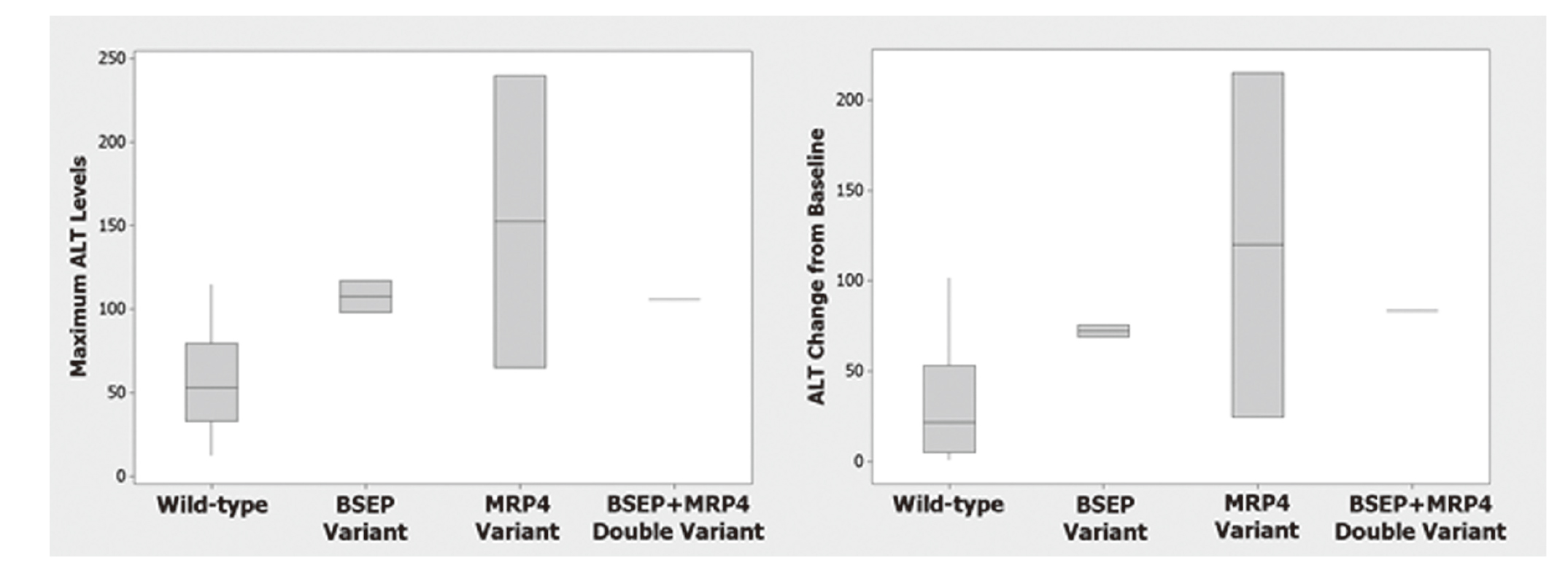
Clinical severity of DILI has been shown to be highly associated with dual inhibition of liver mitochondrial function and BSEP (Aleo et al., 2014). Unfortunately, screening for these two endpoints has not been completely successful in eliminating drug attrition in clinical trials due to liver toxicity, most likely due to the multifactorial nature of clinical DILI. It has been hypothesized that genetic variants in other bile acid transporters [e.g., basolateral transporters such as MRP4 (ABCC4), MRP5 (ABCC5)] and apical transporters such as MDR3 (ABCB4) may be important contributors to DILI risk in addition to BSEP (Mosedale and Watkins, 2017; Sissung et al., 2012). Additionally, AMG009 and TAK875 have been shown to cause DILI via inhibiting multiple bile salt efflux transporters (Morgan et al., 2013; Otieno et al., 2017). Whole exome sequencing was conducted on 30 DNA samples from patients (17 subjects with ALT elevations; 13 without) participating in two Phase 1 clinical trials where liver injury was observed in approximately 57% of patients. Both compounds were BSEP inhibitors, but with 10-fold difference in potency. To investigate which hepatic efflux transporters were associated with DILI risk, therefore guiding the prioritization of additional discovery toxicology screens, linear regression analysis using ALT and exomic profiles from the 30 samples was performed. Despite the limited sample size, this analysis demonstrated that the variants from BSEP or MRP4 were significantly associated with maximum absolute ALT levels and increased ALT levels (Fig. 3). Therefore, MRP4 was prioritized as part of discovery toxicology screens. This example illustrates how human genetic data can be used to inform development of discovery toxicology screens and may potentially improve human translation of these screens.
6-Mercaptopurine (6-MP) is a first line treatment for acute lymphoblastic leukemia and Crohn’s disease (Tzvetkov and von Ahsen, 2012). Leukopenia is the primary adverse drug response that disrupts treatment and the susceptibility to 6-MP differs between European and Japanese ancestry populations. In European-descent patients, thiopurine S-methyltransferase (TPMT) deficiency increases susceptibility to leukopenia where homozygous and heterozygous recessive patients require lower dosages of 6-MP (Blaker et al., 2012). Similarly, nucleoside diphospatase-15 (NUD15) deficiency increases the risk for leukopenia in patients of Japanese descent (Yang et al., 2015). This example illustrates how ancestry-related genetic polymporphisms can mediate adverse drug reactions, and how the field of precision medicine is advancing. For instance, the time from discovery of the biomarker to clinical implementation for TPMT was 21 years compared to 2.5 years for the NUD15 biomarker.
Maraviroc is used in the treatment of HIV, because it inhibits the CCR5 receptor that HIV uses to infect human cells (Dean et al., 1996). The confidence in rationale for this treatment came from the discovery that humans who were homozygous for the Δ32 gene mutation were resistant to HIV infection due to a non-functional CCR5 receptor on the cell surface. Maraviroc is used in patients who have the wild-type receptor or are heterozygous for the Δ32 mutation. Patients who are homozygous for the CCR5Δ32 are excluded from using maraviroc as they would derive no benefit from the drug. This example illustrates how genetic data increases the probability of clinical success and excludes patients who would derive no clinical benefit from its use and only be at risk for side effects.
Cholesterol ester transfer protein inhibitors (CETPi) block transfer of cholesterol into very-low density lipoprotein (VLDL) and thereby increase high-density lipoprotein (HDL) levels. It was believed that increasing HDL would reduce the risk for myocardial infarction and combining CETPi with statins would provide a clinically meaningful benefit. Unfortunately, all attempts to develop CETPi failed in the clinic, including torcetrapid, (2006), dalcetrapib (2012), evacetrapib (2015) and anacetrapib (2017). Using precision medicine principles, variants in the LIPG gene were identified (Voight et al., 2012). Subjects with the LIPG Ser396 variant were determined to have higher HDL-C levels than those with the Asn396 variant. These two variants allowed the study of these two populations, mirroring the CETPi drug intervention trials. The myocardial infarction incidence among these two variant populations was similar which was analogous to the outcome of the CETPi clinical trials. Based on these precision medicine data, it is questionable whether raising HDL cholesterol alone is a viable approach to reducing the risk of myocardial infarction. This example illustrates how clinical failure of the CETPi drugs could have been avoided if precision medicine principles had been applied before choosing to pursue this target, thereby allowing several billion R&D dollars to be invested in other more promising targets.
Phase 4 refers to the post-marketing phase of drug development where health authorities require approved drugs to have a pharmacovigilance plan before launching a drug. In this example, the MOA for statin-induced myopathy was identified as a LOF variant for the SLCO1B1 gene that codes for the OATP1B1 transporter (Ramsey et al., 2014; Tzvetkov and von Ahsen, 2012). A common adverse event for statins is muscle toxicity and it is well recognized that this adverse event is more pronounced with certain statins such as simvastatin. During Phase 4 follow-up, it was demonstrated that statins such as simvastatin exclusively utilized the OATP1B1 transporter for liver uptake and patients were at increased risk for developing muscle toxicity. The increased incidence of muscle toxicity was demonstrated to be associated with subjects who had the LOF variant, rs4149056T>C (Ramsey et al., 2014). Hence, patients who have low OATP1B1 transporter function have higher systemic concentrations of simvastatin, increasing their risk for developing muscle toxicity. This risk is mitigated by using lower doses of simvastatin or prescribing other statins such as atorvastatin, pravastatin, and rosuvastatin that use two additional organic anion transporters, OATP1B3 and OATP2B1, for liver uptake (Tzvetkov and von Ahsen, 2012). This example again illustrates the value in applying precision medicine principles to understand the mechanism of toxicity, and how the results can be used to benefit patients.
SUMMARY AND CONCLUSION
In this review, several examples were used to illustrate why toxicologists should understand precision medicine principles and their application to target selection through Phase 4 clinical development, and how these principles can increase drug survival and decrease the time to regulatory approval. Understanding interindividual differences in response to drugs and how these differences can influence the drug’s risk-benefit profile can result in the discovery of biomarkers that enhance patient safety and efficacy. The discussed examples illustrate why it is important for toxicologists to understand precision medicine principles, and especially in the future, as the field of pharmacogenomics and genetics continues to advance. The development of nonclinical models that can be used to understand genetic variation in humans further underscores the need for toxicologists to understand the principles of precision medicine. It is through the application of precision medicine principles that toxicologists can help improve safety assessments, identify patients with best efficacy/safety profile, reduce pharmaceutical attrition and development time, and facilitate understanding of a drug’s MOA.
ACKNOWLEDGMENTS
The authors thank Dr. Nasir K. Khan for his critical review of this manuscript and Julie Harney and Mark Gosink for help with creating figures presented in this manuscript.
Conflict of interest
All the authors worked for Pfizer Inc. during the conduct of this work.
REFERENCES
- Aleo, M.D., Luo, Y., Swiss, R., Bonin, P.D., Potter, D.M. and Will, Y. (2014): Human drug-induced liver injury severity is highly associated with dual inhibition of liver mitochondrial function and bile salt export pump. Hepatology, 60, 1015-1022.
- An, H.R., Wu, X.Q., Wang, Z.Y., Zhang, J.X. and Liang, Y. (2012): NAT2 and CYP2E1 polymorphisms associated with antituberculosis drug-induced hepatotoxicity in Chinese patients. Clin. Exp. Pharmacol. Physiol., 39, 535-543.
- Blaker, P.A., Arenas-Hernandez, M., Marinaki, A.M. and Sanderson, J.D. (2012): The pharmacogenetic basis of individual variation in thiopurine metabolism. Per. Med., 9, 707-725.
- Bogue, M.A., Churchill, G.A. and Chesler, E.J. (2015): Collaborative Cross and Diversity Outbred data resources in the Mouse Phenome Database. Mamm. Genome, 26, 511-520.
- Church, R.J., Gatti, D.M., Urban, T.J., Long, N., Yang, X., Shi, Q., Eaddy, J.S., Mosedale, M., Ballard, S., Churchill, G.A., Navarro, V., Watkins, P.B., Threadgill, D.W. and Harrill, A.H. (2015): Sensitivity to hepatotoxicity due to epigallocatechin gallate is affected by genetic background in diversity outbred mice. Food Chem. Toxicol., 76, 19-26.
- Church, R.J., Wu, H., Mosedale, M., Sumner, S.J., Pathmasiri, W., Kurtz, C.L., Pletcher, M.T., Eaddy, J.S., Pandher, K., Singer, M., Batheja, A., Watkins, P.B., Adkins, K. and Harrill, A.H. (2014): A systems biology approach utilizing a mouse diversity panel identifies genetic differences influencing isoniazid-induced microvesicular steatosis. Toxicol. Sci., 140, 481-492.
- Court, M.H. (2007): A pharmacogenomics primer. J. Clin. Pharmacol., 47, 1087-1103.
- Court, M.H., Peter, I., Hazarika, S., Vasiadi, M., Greenblatt, D.J. and Lee, W.M.; Acute Liver Failure Study Group. (2014): Candidate gene polymorphisms in patients with acetaminophen-induced acute liver failure. Drug Metab. Dispos., 42, 28-32.
- Dalén, P., Dahl, M.L., Bernal Ruiz, M.L., Nordin, J. and Bertilsson, L. (1998): 10-Hydroxylation of nortriptyline in white persons with 0, 1, 2, 3, and 13 functional CYP2D6 genes. Clin. Pharmacol. Ther., 63, 444-452.
- Dean, M., Carrington, M., Winkler, C., Huttley, G.A., Smith, M.W., Allikmets, R., Goedert, J.J., Buchbinder, S.P., Vittinghoff, E., Gomperts, E., Donfield, S., Vlahov, D., Kaslow, R., Saah, A., Rinaldo, C., Detels, R. and O’Brien, S.J. (1996): Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science, 273, 1856-1862.
- Dolsten, M. and Søgaard, M. (2012): Precision medicine: an approach to R&D for delivering superior medicines to patients. Clin. Transl. Med., 1, 7.
- Ebeling, M., Küng, E., See, A., Broger, C., Steiner, G., Berrera, M., Heckel, T., Iniguez, L., Albert, T., Schmucki, R., Biller, H., Singer, T. and Certa, U. (2011): Genome-based analysis of the nonhuman primate Macaca fascicularis as a model for drug safety assessment. Genome Res., 21, 1746-1756.
- Ewart, L., Dehne, E.M., Fabre, K., Gibbs, S., Hickman, J., Hornberg, E., Ingelman-Sundberg, M., Jang, K.J., Jones, D.R., Lauschke, V.M., Marx, U., Mettetal, J.T., Pointon, A., Williams, D., Zimmermann, W.H. and Newham, P. (2018): Application of Microphysiological Systems to Enhance Safety Assessment in Drug Discovery. Annu. Rev. Pharmacol. Toxicol., 58, 65-82.
- Fattinger, K., Funk, C., Pantze, M., Weber, C., Reichen, J., Stieger, B. and Meier, P.J. (2001): The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: a potential mechanism for hepatic adverse reactions. Clin. Pharmacol. Ther., 69, 223-231.
- Grossman, I. (2009): ADME pharmacogenetics: current practices and future outlook. Expert Opin. Drug Metab. Toxicol., 5, 449-462.
- Harrill, A.H., Watkins, P.B., Su, S., Ross, P.K., Harbourt, D.E., Stylianou, I.M., Boorman, G.A., Russo, M.W., Sackler, R.S., Harris, S.C., Smith, P.C., Tennant, R., Bogue, M., Paigen, K., Harris, C., Contractor, T., Wiltshire, T., Rusyn, I. and Threadgill, D.W. (2009): Mouse population-guided resequencing reveals that variants in CD44 contribute to acetaminophen-induced liver injury in humans. Genome Res., 19, 1507-1515.
- Harrison, R.K. (2016): Phase II and phase III failures: 2013-2015. Nat. Rev. Drug Discov., 15, 817-818.
- Hartman, J.H., Miller, G.P., Caro, A.A., Byrum, S.D., Orr, L.M., Mackintosh, S.G., Tackett, A.J., MacMillan-Crow, L.A., Hallberg, L.M., Ameredes, B.T. and Boysen, G. (2017): 1,3-Butadiene-induced mitochondrial dysfunction is correlated with mitochondrial CYP2E1 activity in Collaborative Cross mice. Toxicology, 378, 114-124.
- Hughes, A.R., Spreen, W.R., Mosteller, M., Warren, L.L., Lai, E.H., Brothers, C.H., Cox, C., Nelsen, A.J., Hughes, S., Thorborn, D.E., Stancil, B., Hetherington, S.V., Burns, D.K. and Roses, A.D. (2008): Pharmacogenetics of hypersensitivity to abacavir: from PGx hypothesis to confirmation to clinical utility. Pharmacogenomics J., 8, 365-374.
- Hustert, E., Haberl, M., Burk, O., Wolbold, R., He, Y.Q., Klein, K., Nuessler, A.C., Neuhaus, P., Klattig, J., Eiselt, R., Koch, I., Zibat, A., Brockmöller, J., Halpert, J.R., Zanger, U.M. and Wojnowski, L. (2001): The genetic determinants of the CYP3A5 polymorphism. Pharmacogenetics, 11, 773-779.
- Ingelman-Sundberg, M., Oscarson, M. and McLellan, R.A. (1999): Polymorphic human cytochrome P450 enzymes: an opportunity for individualized drug treatment. Trends Pharmacol. Sci., 20, 342-349.
- Kuehl, P., Zhang, J., Lin, Y., Lamba, J., Assem, M., Schuetz, J., Watkins, P.B., Daly, A., Wrighton, S.A., Hall, S.D., Maurel, P., Relling, M., Brimer, C., Yasuda, K., Venkataramanan, R., Strom, S., Thummel, K., Boguski, M.S. and Schuetz, E. (2001): Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat. Genet., 27, 383-391.
- Kwak, E.L., Camidge, D.R., Clark, J., Shapiro, G.I., Maki, R.G., Ratain, M.J., Solomon, B., Bang, Y., Ou, S. and Salgia, R. (2009): Clinical activity observed in a phase I dose escalation trial of an oral c-met and ALK inhibitor, PF-02341066. Eur. J. Cancer, Suppl., 7, 8.
- Lang, C., Meier, Y., Stieger, B., Beuers, U., Lang, T., Kerb, R., Kullak-Ublick, G.A., Meier, P.J. and Pauli-Magnus, C. (2007): Mutations and polymorphisms in the bile salt export pump and the multidrug resistance protein 3 associated with drug-induced liver injury. Pharmacogenet. Genomics, 17, 47-60.
- Markova, S.M., De Marco, T., Bendjilali, N., Kobashigawa, E.A., Mefford, J., Sodhi, J., Le, H., Zhang, C., Halladay, J., Rettie, A.E., Khojasteh, C., McGlothlin, D., Wu, A.H., Hsueh, W.C., Witte, J.S., Schwartz, J.B. and Kroetz, D.L. (2013): Association of CYP2C9*2 with bosentan-induced liver injury. Clin. Pharmacol. Ther., 94, 678-686.
- Mas, S. and Lafuente, A. (2009): Pharmacogenetics strategies: from candidate genes to whole-genome association analysis. Exploratory or confirmatory studies? Curr. Pharmacogenomics Person. Med., 7, 59-69.
- Mele, C. and Goldschmidt, K. (2014): Pharmacogenomics in pediatrics: personalized medicine showing eminent promise. J. Pediatr. Nurs., 29, 378-382.
- Moffat, J.G., Vincent, F., Lee, J.A., Eder, J. and Prunotto, M. (2017): Opportunities and challenges in phenotypic drug discovery: an industry perspective. Nat. Rev. Drug Discov., 16, 531-543.
- Morgan, A.P., Fu, C.P., Kao, C.Y., Welsh, C.E., Didion, J.P., Yadgary, L., Hyacinth, L., Ferris, M.T., Bell, T.A., Miller, D.R., Giusti-Rodriguez, P., Nonneman, R.J., Cook, K.D., Whitmire, J.K., Gralinski, L.E., Keller, M., Attie, A.D., Churchill, G.A., Petkov, P., Sullivan, P.F., Brennan, J.R., McMillan, L. and Pardo-Manuel de Villena, F. (2015): The Mouse Universal Genotyping Array: From Substrains to Subspecies. G3 (Bethesda), 6, 263-279.
- Morgan, R.E., van Staden, C.J., Chen, Y., Kalyanaraman, N., Kalanzi, J., Dunn, R.T. 2nd, Afshari, C.A. and Hamadeh, H.K. (2013): A multifactorial approach to hepatobiliary transporter assessment enables improved therapeutic compound development. Toxicol. Sci., 136, 216-241.
- Mosedale, M. (2018): Mouse Population-Based Approaches to Investigate Adverse Drug Reactions. Drug Metab. Dispos. , doi: 10.1124/dmd.118.082834.
- Mosedale, M., Kim, Y., Brock, W.J., Roth, S.E., Wiltshire, T., Eaddy, J.S., Keele, G.R., Corty, R.W., Xie, Y., Valdar, W. and Watkins, P.B. (2017): Editor’s Highlight: Candidate Risk Factors and Mechanisms for Tolvaptan-Induced Liver Injury Are Identified Using a Collaborative Cross Approach. Toxicol. Sci., 156, 438-454.
- Mosedale, M. and Watkins, P.B. (2017): Drug-induced liver injury: advances in mechanistic understanding that will inform risk management. Clin. Pharmacol. Ther., 101, 469-480.
- Mosedale, M., Wu, H., Kurtz, C.L., Schmidt, S.P., Adkins, K. and Harrill, A.H. (2014): Dysregulation of protein degradation pathways may mediate the liver injury and phospholipidosis associated with a cationic amphiphilic antibiotic drug. Toxicol. Appl. Pharmacol., 280, 21-29.
- Nagar, S. and Blanchard, R.L. (2006): Pharmacogenetics of uridine diphosphoglucuronosyltransferase (UGT) 1A family members and its role in patient response to irinotecan. Drug Metab. Rev., 38, 393-409.
- Nelson, M.R., Tipney, H., Painter, J.L., Shen, J., Nicoletti, P., Shen, Y., Floratos, A., Sham, P.C., Li, M.J., Wang, J., Cardon, L.R., Whittaker, J.C. and Sanseau, P. (2015): The support of human genetic evidence for approved drug indications. Nat. Genet., 47, 856-860.
- Otieno, M.A., Bhaskaran, V., Janovitz, E., Callejas, Y., Foster, W.B., Washburn, W., Megill, J.R., Lehman-McKeeman, L. and Gemzik, B. (2017): Mechanisms for Hepatobiliary Toxicity in Rats Treated with an Antagonist of Melanin Concentrating Hormone Receptor 1 (MCHR1). Toxicol. Sci., 155, 379-388.
- Palanisamy, G.S., Marcek, J.M., Cappon, G.D., Whritenour, J., Shaffer, C.L., Brady, J.T. and Houle, C. (2015): Drug-induced Skin Lesions in Cynomolgus Macaques Treated with Metabotropic Glutamate Receptor 5 (mGluR5). Negative Allosteric Modulators. Toxicol. Pathol., 43, 995-1003.
- Pletcher, M.T., Cook, J.C. and Tassinari, M.S. (2010): Individual (Personalized) Vulnerabilities. In: Comprehensive Toxicology Second Edition, Vol. 12, pp. 145-161. Elsevier Inc.
- Qiu, L.O., Linder, M.W., Antonino-Green, D.M. and Valdes, R. Jr. (2004): Suppression of cytochrome P450 2E1 promoter activity by interferon-γ and loss of response due to the -71G>T nucleotide polymorphism of the CYP2E1*7B allele. J. Pharmacol. Exp. Ther., 308, 284-288.
- Ramsey, L.B., Johnson, S.G., Caudle, K.E., Haidar, C.E., Voora, D., Wilke, R.A., Maxwell, W.D., McLeod, H.L., Krauss, R.M., Roden, D.M., Feng, Q., Cooper-DeHoff, R.M., Gong, L., Klein, T.E., Wadelius, M. and Niemi, M. (2014): The clinical pharmacogenetics implementation consortium guideline for SLCO1B1 and simvastatin-induced myopathy: 2014 update. Clin. Pharmacol. Ther., 96, 423-428.
- Rana, S.V., Sharma, S.K., Ola, R.P., Kamboj, J.K., Malik, A., Morya, R.K. and Sinha, S.K. (2014): N-acetyltransferase 2, cytochrome P4502E1 and glutathione S-transferase genotypes in antitubercular treatment-induced hepatotoxicity in North Indians. J. Clin. Pharm. Ther., 39, 91-96.
- Sissung, T.M., Troutman, S.M., Campbell, T.J., Pressler, H.M., Sung, H., Bates, S.E. and Figg, W.D. (2012): Transporter pharmacogenetics: transporter polymorphisms affect normal physiology, diseases, and pharmacotherapy. Discov. Med., 13, 19-34.
- Soda, M., Choi, Y.L., Enomoto, M., Takada, S., Yamashita, Y., Ishikawa, S., Fujiwara, S., Watanabe, H., Kurashina, K., Hatanaka, H., Bando, M., Ohno, S., Ishikawa, Y., Aburatani, H., Niki, T., Sohara, Y., Sugiyama, Y. and Mano, H. (2007): Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature, 448, 561-566.
- Spear, B.B., Heath-Chiozzi, M. and Huff, J. (2001): Clinical application of pharmacogenetics. Trends Mol. Med., 7, 201-204.
- Teixeira, R.L., Morato, R.G., Cabello, P.H., Muniz, L.M., Moreira Ada, S., Kritski, A.L., Mello, F.C., Suffys, P.N., Miranda, A.B. and Santos, A.R. (2011): Genetic polymorphisms of NAT2, CYP2E1 and GST enzymes and the occurrence of antituberculosis drug-induced hepatitis in Brazilian TB patients. Mem. Inst. Oswaldo Cruz, 106, 716-724.
- Threadgill, D.W. and Churchill, G.A. (2012): Ten years of the collaborative cross. G3 (Bethesda), 2, 153-156.
- Turner, R.M. (2013): From the lab to the prescription pad: genetics, CYP450 analysis, and medication response. J. Child Adolesc. Psychiatr. Nurs., 26, 119-123.
- Tzvetkov, M. and von Ahsen, N. (2012): Pharmacogenetic screening for drug therapy: from single gene markers to decision making in the next generation sequencing era. Pathology, 44, 166-180.
- Varmus, H. (2010): Ten years on--the human genome and medicine. N. Engl. J. Med., 362, 2028-2029.
- Vincent, F., Loria, P., Pregel, M., Stanton, R., Kitching, L., Nocka, K., Doyonnas, R., Steppan, C., Gilbert, A., Schroeter, T. and Peakman, M.C. (2015): Developing predictive assays: the phenotypic screening “rule of 3”. Sci. Transl. Med., 7, 293ps15.
- Voight, B.F., Peloso, G.M., Orho-Melander, M., Frikke-Schmidt, R., Barbalic, M., et al. (2012): Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet, 380, 572-580.
- Wang, Y.I., Oleaga, C., Long, C.J., Esch, M.B., McAleer, C.W., Miller, P.G., Hickman, J.J. and Shuler, M.L. (2017): Self-contained, low-cost Body-on-a-Chip systems for drug development. Exp. Biol. Med. (Maywood), 242, 1701-1713.
- Wu, H. and Adkins, K. (2012): Identification of polymorphisms in genes of the immune system in cynomolgus macaques. Mamm. Genome, 23, 467-477.
- Wu, H., Whritenour, J., Sanford, J.C., Houle, C. and Adkins, K.K. (2017): Identification of MHC Haplotypes Associated with Drug-induced Hypersensitivity Reactions in Cynomolgus Monkeys. Toxicol. Pathol., 45, 127-133.
- Yan, G., Zhang, G., Fang, X., Zhang, Y., Li, C., Ling, F., Cooper, D.N., Li, Q., Li, Y., van Gool, A.J., Du, H., Chen, J., Chen, R., Zhang, P., Huang, Z., Thompson, J.R., Meng, Y., Bai, Y., Wang, J., Zhuo, M., Wang, T., Huang, Y., Wei, L., Li, J., Wang, Z., Hu, H., Yang, P., Le, L., Stenson, P.D., Li, B., Liu, X., Ball, E.V., An, N., Huang, Q., Zhang, Y., Fan, W., Zhang, X., Li, Y., Wang, W., Katze, M.G., Su, B., Nielsen, R., Yang, H., Wang, J., Wang, X. and Wang, J. (2011): Genome sequencing and comparison of two nonhuman primate animal models, the cynomolgus and Chinese rhesus macaques. Nat. Biotechnol., 29, 1019-1023.
- Yang, J.J., Landier, W., Yang, W., Liu, C., Hageman, L., Cheng, C., Pei, D., Chen, Y., Crews, K.R., Kornegay, N., Wong, F.L., Evans, W.E., Pui, C.H., Bhatia, S. and Relling, M.V. (2015): Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J. Clin. Oncol., 33, 1235-1242.