Original Article
Thalidomide protects against acute pentylenetetrazol and pilocarpine-induced seizures in mice
2018 Volume 43 Issue 11 Pages 671-684
Details
2018 Volume 43 Issue 11 Pages 671-684
Thalidomide was originally developed to treat primary neurological and psychiatric diseases. There are reports of anticonvulsant effects of thalidomide in rats and antiepileptic effects in patients. Hence, thalidomide (100, 200 and 400 mg/kg) was herein administered to mice to evaluate possible protection against seizures induced by the systemic administration of neurotoxins: 10 mg/kg of 4-aminopyridine (4-AP), 90 mg/kg of pentylenetetrazol (PTZ), or 380 mg/kg of pilocarpine. The effect of an NO and COX inhibitor (7-NI and ibuprofen, respectively) was also examined. The results show that thalidomide (1) induces the typical sedative effects, (2) has no anticonvulsant effect in mice treated with 4-AP, and (3) has anticonvulsant effect (400 mg/kg) in mice treated with PTZ and pilocarpine. It was found that 7-NI has an anticonvulsant effect in the pilocarpine model and that thalidomide’s effect is not enhanced by its presence. However, thalidomide (200 mg/kg) plus 7-NI or ibuprofen tend to have a toxic effect in PTZ model. On the other hand, the combination of thalidomide and 7-NI or ibuprofen protects against pilocarpine-induced seizures. In conclusion, thalidomide did not exert an anticonvulsant effect for clonic-tonic type convulsions (4-AP), but it did so for seizures induced by PTZ and pilocarpine (representing absence seizures and status epilepticus, respectively). NO and prostaglandins were involved in the convulsive process elicited by pilocarpine.
Epilepsy is a chronic neurological disorder characterized by recurrent seizures, which can vary from brief episodes of absence or muscle spasms to severe and prolonged seizures (Fisher et al., 2014). According to statistics from the WHO, there are about 50 million people suffering from epilepsy in the world, approximately 80% of whom live in developing countries. One third of epileptics do not respond to the pharmacological treatments (WHO, 2017). There is evidence that inflammation takes part in seizures disorders with different etiology. Some of the studies have demonstrated an important activation of the inflammatory cytokine network in patients with refractory epilepsy (Vezzani et al., 2013). Due to that, it is important to study drugs with dual mechanisms, such as thalidomide.
Thalidomide was introduced in the 1950s as a safe antiepileptic drug. In 1957, it was commercialized as a safe sedative and was widely used as an antiemetic (Randall, 1990; Perri and Hsu, 2003). In 1961, however, it was withdrawn from the market because of its teratogenic effects (Hashimoto, 1998). Afterwards, other outstanding pharmacological properties were discovered (Palencia et al., 2007).
Some studies have found a decrease in the frequency and intensity of seizures due to the administration of thalidomide to patients with Rasmussen encephalitis. These results were attributed to the immunomodulatory properties of thalidomide rather than to its direct effect on the central nervous system (Marjanovic et al., 2003). In 2010, Palencia et al. documented outstanding therapeutic effects produced by thalidomide in refractory epilepsy. In experimental models, a notable dose-dependent anticonvulsant effect of thalidomide has been observed in rats exposed to pentylenetetrazol (PTZ; Palencia et al., 2007). Later, Palencia et al. (2011) showed that low doses of thalidomide exhibit anticonvulsant properties in rats subjected to the amygdala kindling model.
Although the mechanisms of action of thalidomide are not clearly understood, it is known to act by modulating different cytokines, including TNF-α, interleukins 10 and 12, γ interferon, cyclooxygenases (COX) and NO (Noguchi et al., 2002; Suizu et al., 2003; Franks et al., 2004; Payandemehr et al., 2014). Therefore, thalidomide is classified as a multi-target drug (Shimazawa et al., 2004).
Two pharmacological interactions were explored by employing an nNOS inhibitor and a COX inhibitor. COXs play a role in the pathogenesis of epilepsy, mainly due to the production of proinflammatory prostaglandins. In this sense, there is evidence that seizure activity increases the levels of COX-2 (Dhir et al., 2006) and NO during the development and maintenance of inflammation (Saad et al., 2016). Rajasekaran et al. (2003) detected greater levels of neuronal nitric oxide synthase (nNOS) in several regions of the rat brain during the period of seizures.
The first approach to find a potential anti-seizure drug is to test it against different acute models of convulsions, that are related with the physiopathological mechanisms of the seizures in humans (Rubio et al., 2010). The most common and simple seizure models are the pentylenetetrazole (PTZ) and maximal electroshock models (Löscher, 2011); the second one could be substituted with the 4-aminopyridine (4-AP) chemical agent (Yamaguchi and Rogawski, 1992). PTZ and 4-AP are neurotoxins. PTZ induces seizures by the blockade of the GABAA channel in an allosteric site (Rubio et al., 2010), while the 4-AP blocks the action of the voltage-dependent K+ channels in the terminals, causing an increase in the neurotransmitter release (Yamaguchi and Rogawski, 1992). Moreover, the pilocarpine acute model has been less studied, but is a model for pharmacoresistant seizures (Löscher, 2011). Pilocarpine induces status epilepticus by the activation of the M1 muscarinic receptors, mainly in the septum; the activation causes an increase of glutamate level in the hippocampus that onset the seizures (Curia et al., 2008).
To complement the reports in preclinical research of the anticonvulsant effects of thalidomide, the aim of the present study was to systematically evaluate its anticonvulsant and indirectly the anti-inflammatory properties in three essential models of seizures: 4-AP, PTZ and pilocarpine in mice, including the combination of thalidomide with a selective nNOS inhibitor or a COX inhibitor, expecting to find less inflammation and thus reduced seizure activity.
Male CD1 mice were obtained from the animal facility No. 1 (room 3) of the Physiology Department of the Escuela Nacional de Ciencias Biologicas of the Instituto Politecnico Nacional. Harlan Laboratories was the original animal source for the animal breeding. Male CD1 mice weighing 25-30 g were kept in a room under standard conditions (22 ± 2°C, 55 ± 5% humidity and a 12:12 hr light/dark cycle) and provided food and water ad libitum. For all the experiments performed in the study, the groups were formed by 12 mice. All experiments and manipulations were performed in accordance with the guidelines for the Use and Care of Laboratory Animals (NOM-062-ZOO-1999, NOM-033-ZOO-1995 and NOM-029-ZOO-1995), and the protocol was approved by the institutional committee for animal use.
DrugsThe drugs used in the study were: thalidomide, 7-NI (a neuronal NOS inhibitor) and ibuprofen (a COX inhibitor) obtained from Cayman Chemical, Ann Arbor, MI, USA; 4-AP, PTZ, pilocarpine, scopolamine methyl bromide and SVP from Sigma-Aldrich, St. Louis, MO, USA; diazepam (DZP) from Cryopharma Laboratories, Tlajomulco de Zuñiga, Jalisco, Mexico.
Different doses of thalidomide were suspended in PBS with 0.5% carboxymethylcellulose (Sigma-Aldrich), while 7-NI was suspended in an aqueous solution of 1% Tween 80, and ibuprofen in an aqueous solution of 5% DMSO. The rest of the drugs were dissolved in physiological saline solution (NaCl 0.9%).
Locomotor activity testFour groups of mice were formed: one group received the vehicle and three groups were given distinct doses of thalidomide (100, 200 or 400 mg/kg). Treatments were administered i.p. and the open field test was performed one hour later. It consisted of placing each mouse in the center of a box (60 x 60 x 40 cm) and then observing its activity for 5 min to determine the time of ambulation, stereotypy and immobility (Brenes Sáenz et al., 2006; Gould et al., 2009). Behavior was quantified with the JWatcher program.
Acute models of seizuresThe anticonvulsant properties of thalidomide were evaluated subsequent to i.p. administration in mice at doses of 100, 200 or 400 mg/kg (n = 12). SVP was used as the reference drug at a dose of 400 mg/kg for the 4-AP model and 300 mg/kg for the PTZ model. For the pilocarpine model, diazepam at 10 mg/kg was the reference.
4-Aminopyridine modelOne hour after the treatments (30 min in the case of the positive control), the convulsant agent (4-AP 10 mg/kg) was administered subcutaneously and the animals were observed for 1 hr (Yamaguchi and Rogawski, 1992). The parameters measured in the models were: latency to convulsions, number of convulsions, total duration of convulsions, latency to death, and convulsive and death rate.
Pentylenetetrazole modelThe procedure was similar to the 4-AP model, except that animals were observed for 30 min and PTZ administered at a dose of 90 mg/kg (Kasture et al., 2002).
Pilocarpine modelForty-five min after administering the aforementioned treatments in the negative control and the thalidomide groups, scopolamine methyl bromide (1 mg/kg) was applied to block the peripheral cholinergic effects (Khongsombat et al., 2008) of pilocarpine (380 mg/kg), which was administered 15 min later. The positive control group received scopolamine methyl bromide, followed by DZP 10 min later and then pilocarpine 5 min afterwards. All mice were observed for two hours.
The parameters measured in the model were: latency to convulsions, number of convulsions, total duration of convulsions, latency to death, and convulsive and death rate. Additionally, the maximum stage of neuronal excitability was assessed (Racine’s scale modified by Borges et al., 2003).
Anticonvulsant properties of thalidomide in the presence of an nNOS inhibitorDue to the previous results, the 200 mg/kg dose of thalidomide was chosen for the assay with 7-NI as an nNOS inhibitor (at 20 mg/kg). The anticonvulsant properties of thalidomide were evaluated in the aforementioned 3 acute seizure models, forming four groups (12 mice each) per model: the control group (PBS and 1% aqueous solution of Tween 80), the thalidomide group, the 7-NI group, and the 7-NI plus thalidomide group. The convulsant agents and the time of observation varied as appropriate. Thalidomide was administered at time zero, 7-NI at 15 and at 60 the convulsant drug depending on the model.
Anticonvulsant properties of thalidomide in the presence of a COX inhibitorThe dose of thalidomide was 200 mg/kg in the assay with ibuprofen as a non-selective COX inhibitor (at 100 mg/kg; Saad et al., 2016). Thalidomide was administered at time zero and ibuprofen 15 min later. One hour after injecting thalidomide, the convulsant drug (4-AP, PTZ or pilocarpine) was given.
Statistical analysesThe results are expressed as the mean ± S.E. for parametric data, and as the median of values having a range between the 5th and 95th percentile for non-parametric data. Statistical significance was determined with one-way ANOVA or the Kruskal-Wallis test, followed (if applicable) by the Student-Newman-Keuls test. For nominal data (frequencies), Fisher’s exact probability test or chi-square test was used. Differences were considered significant with a P-value < 0.05. Statistical analysis was run on Sigma Plot 12.0.
The results from the open field test (Fig. 1) revealed the sedative activity of thalidomide in mice, evidenced by a significant reduction in the ambulatory and stereotypy time of mice (with all three doses of the drug), and a significant increase in resting time.
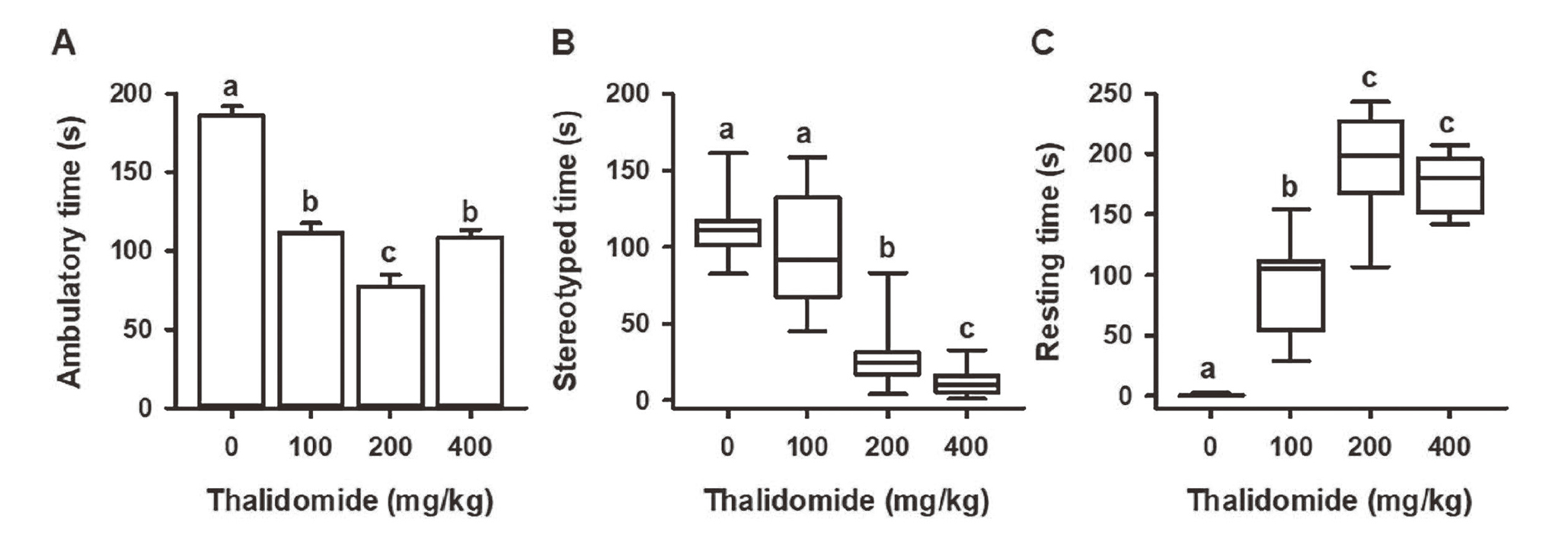
Effect of thalidomide on spontaneous motor activity: A) ambulatory time (sec), B) stereotyped time (sec) and C) resting time (sec). Values represent the mean ± S.E. or median ± P5 and P95. The different lowercase letters indicate P < 0.05.
Specifically, a significant dose-dependent decrease (versus the control) was observed in ambulatory time (Fig. 1A) for animals administered thalidomide at doses of 100 and 200 mg/kg. There was also a shorter ambulatory time with the highest dose of thalidomide (compared to the control), but the effect was less than that produced by 200 mg/kg. Similarly, the maximum value for resting time was obtained at the doses of 200 and 400 mg/kg (Fig. 1C). Meanwhile, mice given 400 mg/kg showed a significant decline in stereotypy time (Fig. 1B), and all results were dose dependent.
Protection against 4-aminopyridine-induced seizuresThe method was validated by the significant differences found for every evaluated parameter when comparing the negative (vehicle) and positive (sodium valproate: SVP) control. At the dose of 200 mg/kg of thalidomide, the latency to convulsions was significantly longer (Fig. 2B) and the duration of convulsions significantly shorter (Fig. 2D). The latter parameter was unchanged at the highest dose of thalidomide. No statistical differences were detected in regard to the convulsive rate (Fig. 2A), number of convulsions (Fig. 2C) or death rate (Fig. 2F) when comparing any of the doses of thalidomide to the control.

Effect of thalidomide on the parameters of convulsions and death in the model of 4-AP-induced seizures: A) convulsive rate (%), B) latency to convulsions (min), C) number of convulsions (n), D) total duration of convulsions (sec), E) latency to death (min) and F) death rate (%). Values represent the mean ± S.E. or median ± P5 and P95. The different lowercase letters indicate P < 0.05.
In this model, thalidomide showed protection against convulsions (Fig. 3A) and death (Fig. 3F) at 400 mg/kg. Compared to the control, significant increases or decreases were found for different parameters when thalidomide was administered at the doses of 200 mg/kg and 400 mg/kg. There was no significant difference for almost any evaluated variable when comparing thalidomide at 400 mg/kg and SVP (Fig. 3). The 200 mg/kg dose of thalidomide is considered the threshold dose for the anticonvulsant effect.

Effect of thalidomide on the parameters of convulsions and death in the model of PTZ-induced seizures: A) convulsive rate (%), B) latency to convulsions (min), C) number of convulsions (n), D) total duration of convulsions (sec), E) latency to death (min) and F) death rate (%). Values represent the mean ± S.E. or median ± P5 and P95. The different lowercase letters denote P < 0.05.
An anticonvulsive effect of thalidomide was observed for status epilepticus with the highest dose of the drug (Fig. 4). Only mice given 400 mg/kg of thalidomide showed favorable significant differences in the convulsive rate (Fig. 4A), latency to convulsions (Fig. 4B) and death (Fig. 4F), death rate (Fig. 4G) and maximum stage of neuronal excitability (Fig. 4E).

Effect of thalidomide on parameters of convulsions and death in the model of pilocarpine-induced seizures: A) convulsive rate (%), B) latency to convulsions (min), C) number of convulsions (n), D) total duration of convulsions (sec), E) maximum stage of neuronal excitability, F) latency to death (min) and G) death rate (%). Values represent the mean ± S.E. or median ± P5 and P95. The different lowercase letters indicate P < 0.05.
In the 4-AP model, no significant differences existed between groups (Fig. 5). In the PTZ model, the number of convulsions was significantly lower (versus the control) in the group receiving the combination of thalidomide (200 mg/kg) and 7-nitroindazole (7-NI; 20 mg/kg) (Fig. 6C), and the total duration of convulsions was significantly reduced (Fig. 6D) in the groups treated with thalidomide (200 mg/kg), 7-NI (20 mg/kg), and the combination of both drugs. No significant difference was detected between the three treatments compared with the control, when the death rate was evaluated (Fig. 6F). However, the death rate was increased in the 7-NI plus thalidomide group, whereas it was decreased in the thalidomide and 7- NI groups, but both effects do not differ compared to the control; the reason why is that the combination of both treatments significantly differs from the two groups.

Effect of thalidomide and 7-nitroindazole on the parameters of convulsions and death in the model of 4-AP-induced seizures: A) convulsive rate (%), B) latency to convulsions (min), C) number of convulsions (n), D) total duration of convulsions (sec), E) latency to death (min) and F) death rate (%). The values represent the mean ± S.E. or median ± P5 and P95. The different lowercase letters indicate P < 0.05.

Effect of thalidomide and 7-nitroindazole on the parameters of convulsions and death in the model of PTZ-induced seizures: A) convulsive rate (%), B) latency to convulsions (min), C) number of convulsions (n), D) total duration of convulsions (sec), E) latency to death (min) and F) death rate (%). The values represent the mean ± S.E. or median ± P5 and P95. The different lowercase letters denote P < 0.05.
In the pilocarpine model (Fig. 7), mice treated with 7-NI and the combination of thalidomide and 7-NI were provided protection (compared to the control) against seizures (Fig. 7A) and death (Fig. 7G). Such protection included significantly longer latency to convulsions and latency to death. Similarly, the number of convulsions, the total duration of convulsions and the maximum stage of excitability were significantly lower in almost every treated group compared to the control.

Effect of thalidomide and 7-nitroindazole on the parameters of convulsions and death in the model of pilocarpine-induced seizures: A) convulsive rate (%), B) latency to convulsions (min), C) number of convulsions (n), D) total duration of convulsions (sec), E) maximum stage of neuronal excitability, F) latency to death (min) and G) death rate (%). The values represent the mean ± S.E. or median ± P5 and P95. The different lowercase letters indicate P < 0.05.
In the 4-AP model, no significant differences existed between groups (Fig. 8). In the PTZ model, the number of convulsions (Fig. 9C) and the total duration of convulsions (Fig. 9D) were significantly lower (versus the control) with thalidomide administered alone at 200 mg/kg and thalidomide plus ibuprofen. Protection against death was found only with the administration of thalidomide alone at 200 mg/kg (Figs. 9E, 9F).

Effect of thalidomide and ibuprofen on the parameters of convulsions and death in the model of 4-AP-induced seizures: A) convulsive rate (%), B) latency to convulsions (min), C) number of convulsions (n), D) total duration of convulsions (sec), E) latency to death (min) and F) death rate (%). The values represent the mean ± S.E. or median ± P5 and P95. The different lowercase letters indicate P < 0.05.
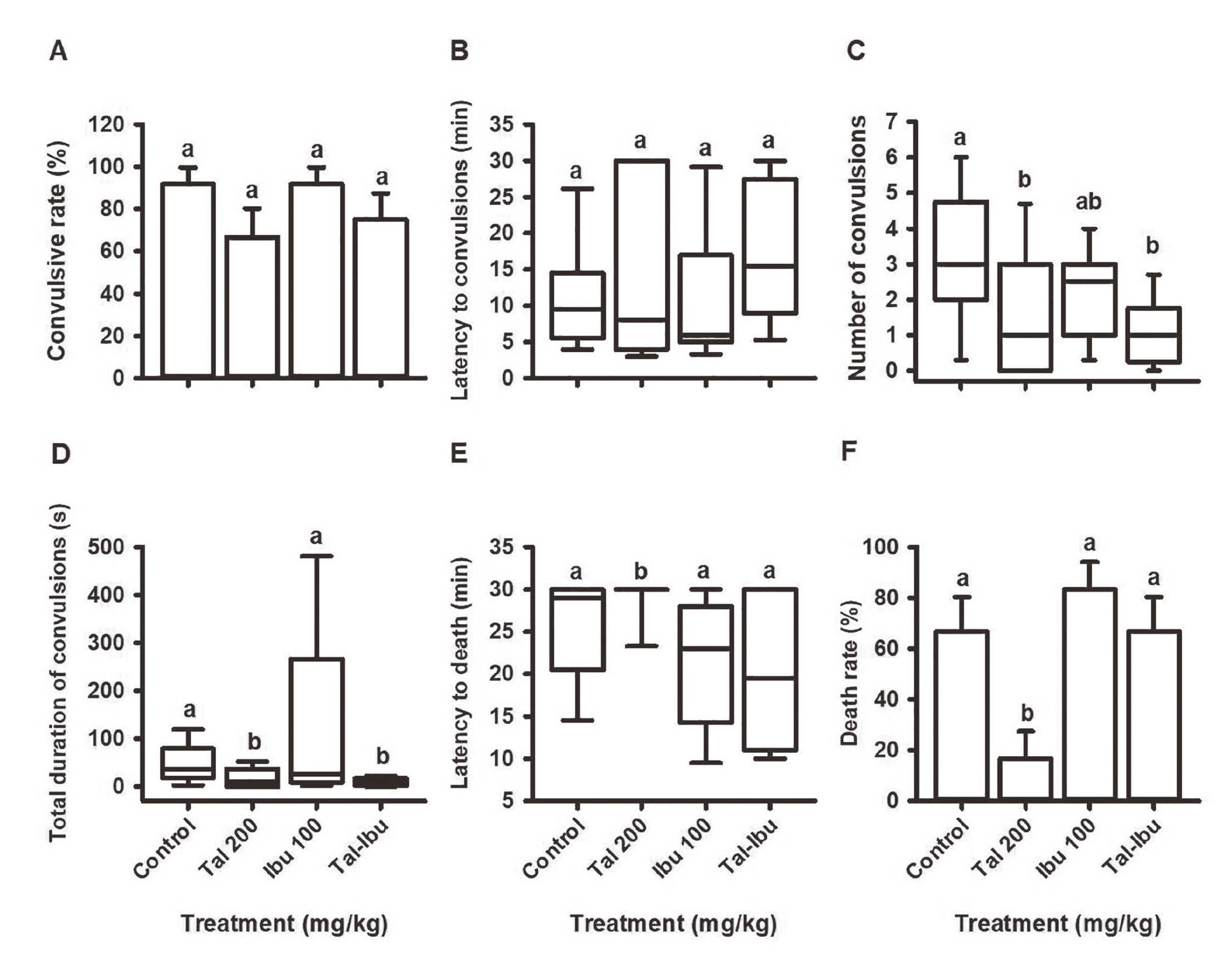
Effect of thalidomide and ibuprofen on the parameters of convulsions and death in the model of PTZ-induced seizures: A) convulsive rate (%), B) latency to convulsions (min), C) number of convulsions (n), D) total duration of convulsions (sec), E) latency to death (min) and F) death rate (%). The values represent the mean ± S.E. or median ± P5 and P95. The different lowercase letters denote P < 0.05.
Compared to the control group, only the combination of thalidomide (200 mg/kg) plus ibuprofen (100 mg/kg) resulted in protection against seizures and death in the pilocarpine model (Fig. 10), which was evidenced by a significant difference in every single evaluated parameter. The administration of thalidomide (200 mg/kg) or ibuprofen (100 mg/kg) alone reduced (versus the control) the number of convulsions (Fig. 10C) and the total duration of convulsions (Fig. 10D). However, the effect of each drug alone was not significantly higher than that found with the combination of thalidomide and ibuprofen.
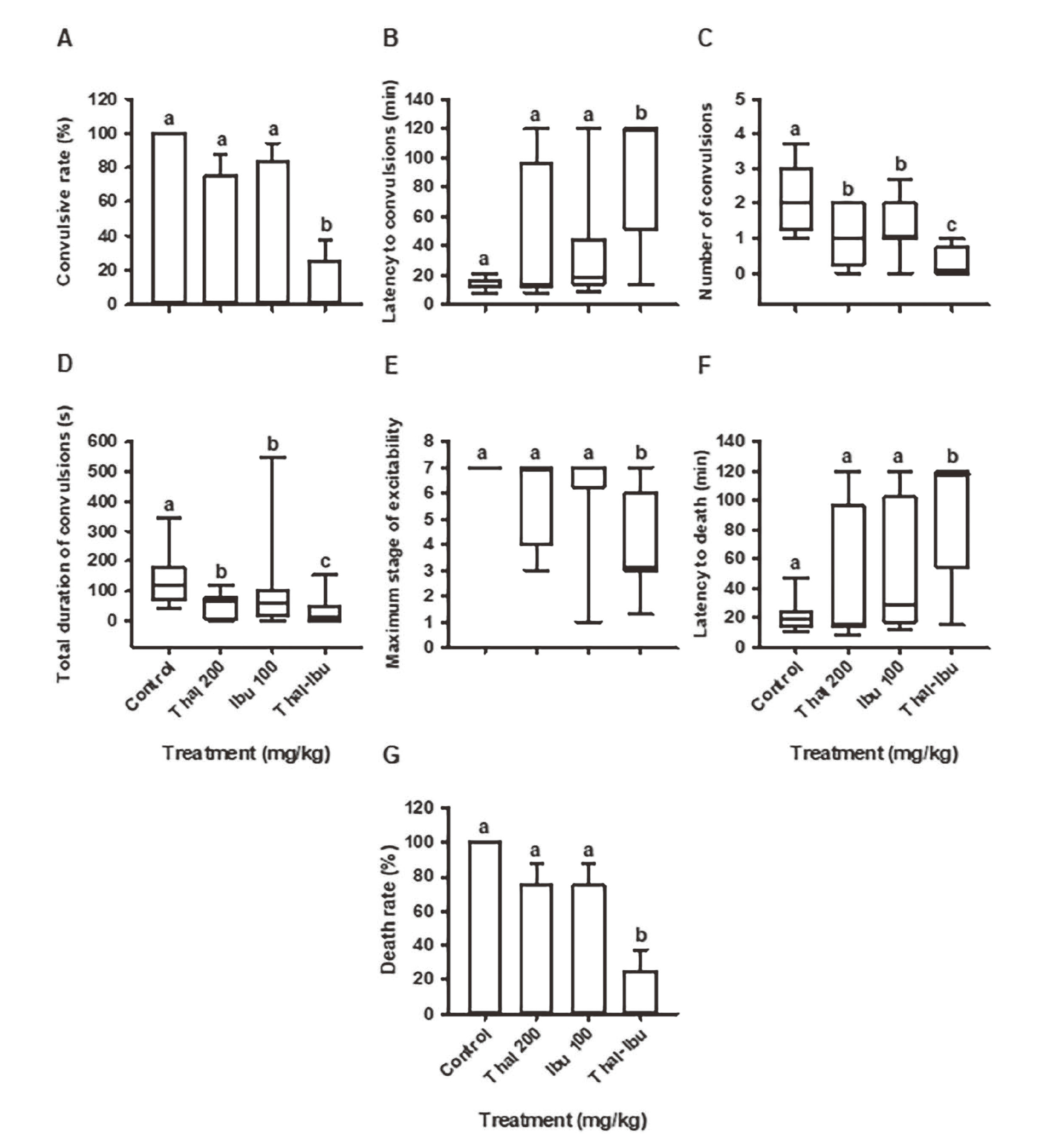
Effect of thalidomide and ibuprofen on the parameters of convulsions and death in the model of pilocarpine-induced seizures: A) convulsive rate (%), B) latency to convulsions (min), C) number of convulsions (n), D) total duration of convulsions (sec), E) maximum stage of neuronal excitability, F) latency to death (min) and G) death rate (%). The values represent the mean ± S.E. or median ± P5 and P95. The different lowercase letters indicate P < 0.05.
After analyzing its pharmacological properties in mice in 1960, Somers determined that thalidomide has a calming effect on the central nervous system, reducing the voluntary activity of the animals and promoting sleep. The current results verify the previously found sedative activity of thalidomide.
In the 4-AP model, thalidomide did not elicit an anticonvulsant effect in mice at any of the test doses. However, thalidomide at 200 mg/kg produced a significantly greater latency to convulsions and a shorter total duration of convulsions, which could be interpreted as marginal protection. The effect of 4-AP is caused by blocking transient and sustained voltage-dependent K+ channels, leading to generalized clonic-tonic convulsions (Yamaguchi and Rogawski, 1992) so intense that none of the present thalidomide doses was able to protect mice from them. Therefore, thalidomide would not be effective against maximal electroshock (Cramer et al., 1994). It is clear that two lower doses of thalidomide (100 and 200 mg/kg) do not protect against 4-AP. Nevertheless, it seemed like an “additive effect” occurred between thalidomide at 400 mg/kg and the convulsant drug (4-AP): the latency to convulsions (Fig 2B) and the latency to death (Fig. 2E) significantly decreased compared to the control and the other tested treatments. Also, mice treated with the highest dose of thalidomide, suffered from one seizure with enough intensity to kill them almost immediately. So far, we do not have an explanation for such observed effect; probably thalidomide begins to be toxic at that dose in combination with the convulsant agent.
The anticonvulsant effects of thalidomide in the mouse PTZ model of the current contribution are similar to the dose-dependent effects observed by Palencia et al. (2007) in rats exposed to the same convulsant agent. It is important to note that the minimum effective dose of thalidomide in mice is 200 mg/kg, while being 25 mg/kg in rats. In either case, a drug that can block pentylenetetrazol may potentially be used for the treatment of absence seizures (De Deyn et al., 1992). Additionally, the protection afforded by thalidomide against PTZ-induced seizures offers a clue as to a possible mechanism of action. To provide this protection, thalidomide might directly interact with the GABAergic system by antagonizing the effects of PTZ at the allosteric site, acting as a barbiturate or benzodiazepine, inhibiting the metabolism of GABA or inhibiting the GABA transporter. Moreover, it might be a calcium channel blocker as ethosuximide.
Following the binding of pilocarpine to M1 receptors, seizures may be sustained by the activation of NMDA receptors (Smolders et al., 1997), causing an imbalance in the neurotransmission that leads in excitotoxicity. The consequent increase in calcium concentration activates the COXs, leading to inflammation in the brain. The protection furnished presently by thalidomide in the pilocarpine model is likely due to its anti-inflammatory activity, which has been demonstrated previously (Barnhill et al., 1984; Lima et al., 2002; Noguchi et al., 2002) and the enhacement of the inhibitory neurotransmition action could be added (Palencia et al., 2007).
NO is another factor to be considered, as it probably mediates some pathophysiological aspects of seizures and seems to be an end product of many excitatory pathways involved in seizures (Bahremand et al., 2010). It is well known that the excitotoxic glutamate activity in the NMDA receptors promotes an increase in the NO synthesis by the over-activation of the nNOS, leading an excitotoxic mechanism mediated by NO (Brown, 2010). Otherwise, there is some evidence supporting the presence of nNOS in hippocampal GABAergic neurons, where in physiological concentration of NO potentiates the inhibitory transmission (Zanelli et al., 2009).
By generating seizures with the three convulsant agents presently employed, we expected an increase in the concentration of NO. Hence, the administration of a NO neuronal synthase inhibitor was included in the study to provide a protective effect against seizures and death elicited by NO. An even greater protection should then result from combining this NO inhibitor with thalidomide, but in some cases the combinations causes a tendency to increase the death rate (Fig. 6F). The phenomena may be explained due to an over-inhibition of the nNOS by thalidomide and 7-NI, limiting the NO synthesis to potentiate the GABAergic transmission in the inhibitory synapsis, that could be translate in the maintenance of the over-activation of the excitatory transmission producing a severe neurotoxic damage. The expected protection was only observed in the pilocarpine model, which at least confirmed that NO synthase inhibitors can protect against seizures (Osonoe et al., 1994). Apparently, such inhibitors protect only against certain types of seizures. The activity of 7-NI likely impeded the elevation of NO levels in the brain, and therefore inflammation was not favored. The per se anti-inflammatory activity of thalidomide probably also countered inflammation in the brain induced by pilocarpine.
To explore another anti-inflammatory pathway, assays were performed with ibuprofen (alone and in combination with thalidomide), a non-selective COX inhibitor. In the 4-AP, the mechanisms of thalidomide and ibuprofen are still not effective to reduce the toxicity evoked by the neurotoxin. Speaking about the PTZ experiment, it was possible to observe the tendency of thalidomide and the combination of thalidomide and ibuprofen to protect, nonetheless the combination of the drugs tends to increase the death rate (Fig. 9F). The lack of the effect could be addressed to the bidirectional effect of the COX-2 in epilepsy. COX-2 may be a neuroprotective agent in the early stage of developing seizures (Shimada et al., 2014). The acute PTZ-induced seizure model has a very short period of time to evaluate the seizure progression and the generation of the seizure activity due to the administration of a high dose of the neurotoxin. It is possible than in 30 min, the ibuprofen inhibits the COX-2 neuroprotective phase in the seizure development that leads in a severe neuronal damage, consequently the mice died (Rojas et al., 2014) and the thalidomide threshold dose is not enough to balance the transmission. It is known that PTZ boosts the levels of PGF2α, PGE2 and TXB2 in the mouse brain (Steinhauer and Hertting, 1981). With this in mind, it makes sense that the anti-inflammatory activity of thalidomide was herein improved by administering it with ibuprofen, leading to a positive impact on some of the parameters evaluated in the PTZ model. Ibuprofen does not have effect on thalidomide’s action against convulsions in the PTZ model (Fig. 9A), but it does enhanced protection against convulsions in the pilocarpine model (Fig. 10A), these results mean ibuprofen potentiates the anticonvulsant action of thalidomide just in the pilocarpine model, because of its anti-inflammatory properties, highlighted in this model.
When mice were pretreated with the combination of thalidomide and ibuprofen, protection against pilocarpine-induced seizures was significantly enhanced. A remarkable finding in the study was that thalidomide and ibuprofen alone tend to reduce the number and duration of seizures, however their mechanisms of action are not enough to attenuate the epileptic phenomena. When mice were treated in combination with both drugs, the anticonvulsant effect is significant and might be explain due to the thalidomide enhancing effect over the inhibitory neurotransmission and the anti-inflammatory effect, summing up the non-selective anti-inflammatory effect of ibuprofen in a late phase of seizure onset, where the COX plays an important role in epilepsy development. Our results are in agreement with reports on the decrease in seizures with some anti-inflammatory treatments, both in experimental models and clinical cases of epilepsy (Vezzani and Granata, 2005). After inducing status epilepticus with pilocarpine in rats, Naffah-Mazzacoratti et al. (1995) found greater levels of PGF2α, PGE2 and PGD2 in the hippocampus. Furthermore, the level of each prostaglandin increased in accordance with the length of time of the pilocarpine model.
It is clear that COX and prostaglandins play an important role in susceptibility to seizures. In 2003, Takemiya et al. examined the effects of COX-2 on a rapid kindling mouse model. They determined that PGE2 is mostly produced by COX-2, after seizure. Additionally, this prostaglandin facilitates neural synaptic excitability by stimulating the release of glutamate from the nerve terminals. In addition, it has been demonstrated that inflammation plays an important role in temporal lobe epilepsy models, such as the induced by pilocarpine (Voutsinos-Porche et al., 2004; Auvin et al., 2010).
In conclusion, the current findings demonstrate that thalidomide does not protect the mice from the neurotoxic effect of 4-aminopyridine. In PTZ and pilocarpine-induced seizures, thalidomide elicited an anticonvulsant effect at 400 mg/kg. Moreover, the combination of thalidomide 200 mg/kg with 7-NI or ibuprofen tends to protect against PTZ-induced seizures, but they triggers a toxic effect that increasd the death of mice; whereas in the pilocarpine model, the combination of the drugs have a clearly anticonvulsant effect. Apparently, NO and COX-2 play an important role in the inmflammation produced by the seizures in the pilocarpine model, which mimics the TLE.
We are grateful for the funds provided by the Instituto Politécnico Nacional (SIP: 20161041). AMI and CC are fellows of the Consejo Nacional de Ciencia y Tecnología (576578 and 275849, respectively). ER is fellow of SNI. Authors thank Bruce Allan Larsen for English language assistance.
Conflict of interestThe authors declare that there is no conflict of interest.