Original Article
Monomethylmercury degradation by the human gut microbiota is stimulated by protein amendments
2018 Volume 43 Issue 12 Pages 717-725
Details
2018 Volume 43 Issue 12 Pages 717-725
Monomethylmercury (MMHg) is a potent neurotoxicant that can be bioaccumulated and biomagnified through trophic levels. Human populations whose diets contain MMHg are at risk of MMHg toxicity. The gut microbiota was identified as a potential factor causing variation in MMHg absorption and body burden. However, little is known about the role of gut microbiota on Hg transformations. We conducted a series of in vitro experiments to study the effects of dietary nutrient change on Hg metabolism and the human gut microbiota using anoxic fecal slurry incubations. We used stable Hg isotope tracers to track MMHg production and degradation and characterized the microbiota using high throughput sequencing of the 16S rRNA gene. We show that the magnitude of MMHg degradation is individual dependent and rapidly responds to changes in nutrient amendments, leading to complete degradation of the MMHg present. Although the mechanism involved remains unknown, it does not appear to involve the well-known mer operon. Our data are the first to show a nutrient dependency on the ability of the simulated human gut microbiota to demethylate MMHg. This work provides much-needed insights into individual variations in Hg absorption and potential toxicity.
Global mercury (Hg) pollution leads to an increase in monomethylmercury (MMHg) levels in fish and seafood and poses a health risk to human consumers (Mergler et al., 2007). The World Health Organization and the Food and Agriculture Organization recommend the consumption of fish as its health benefits outweigh the potential risk associated with MMHg toxicity (Food and Agriculture Organization of the United Nations and World Health Organization, 2011; Starling et al., 2015). To set these guidelines, the body burden of MMHg is often estimated by measuring Hg concentration in hair. However, there is considerable variation between the estimated MMHg oral intake and hair Hg concentrations in most populations tested (Canuel et al., 2006). To estimate human exposure to MMHg, risk assessors often use standard models that assume that 90 to 100% of MMHg (Aberg et al., 1969) and 7-15% of Hg (Miettinen, 1973) ingested is bioavailable. However, experimental evidence suggests that absorption estimates range from 12% to 79% for MMHg and 49% to 69% for inorganic divalent (HgII) (Bradley et al., 2017). This discrepancy points to the existence of unidentified factors that are currently not considered in modelling the oral intake and body burden of MMHg.
The mechanism of absorption of MMHg from the intestine has been well documented (Naganuma et al., 1991). MMHg has a much higher absorption rate than HgII (Bridges and Zalups, 2010). Therefore, the demethylation of MMHg in the gastrointestinal tract will decrease its absorption. The origin of the food, cooking methods and co-ingested micronutrients (e.g., selenium, fruit fibres) can potentially alleviate MMHg toxicity by reducing its bioavailability and bioaccessibility (Passos et al., 2007; Ralston and Raymond, 2010; Ouédraogo and Amyot, 2011). Li et al. (2016) have shown that age, body weight, gender, ethnicity, household income, and geographic regions only account for 32% of the total variance measured in hair Hg concentrations. The authors suggested that other factors such as diet or the activity of the gut microbiota might explain the remaining variance. Rand et al. (2016) reported a high degree of inter-individual variability in the MMHg biotransformation kinetics during a feeding experiment and proposed that the gut microbiome can be a significant modulating factor for MMHg absorption. Later, Caito et al. (2018) showed that individuals who received antibiotic therapy had lower MMHg elimination rates than untreated individuals, highlighting the importance of gut microbiota in the elimination of MMHg.
Hg biotransformation in the gut environment is largely unexplored. Few studies have reported that the gut of fish and zooplankton can potentially methylate Hg (Wang et al., 2013; Gorokhova et al., 2018) and the gut of rat and marine fish can demethylate MMHg (Nakamura et al., 1977; Rowland et al., 1984; Urano et al., 1990; Wang et al., 2017). The human gut does not appear to be a hotspot for MMHg production (Podar et al., 2015) but appears suitable for MMHg degradation (Rowland, 1988). Genes involved in Hg methylation (hgcA and hgcB) were not detected in the gut microbiome of healthy human individuals (Gilmour et al., 2013; Podar et al., 2015). However, genes involved in MMHg demethylation, encoded by the mer operon were detected (Osborn et al., 1997; Rothenberg et al., 2016).
There is a knowledge gap on the genetic determinants and microbial mechanisms involved in the biotransformation of Hg in the gut environment. Although a great emphasis has been placed on the mechanisms that control the formation of MMHg in sediments and soils, fewer studies have addressed the fate of MMHg after it is produced. Most of our knowledge of MMHg biotransformation was gathered from experiments performed outside the human gut (e.g., lake sediments, wetlands). Two pathways can usually be identified for bacterially mediated MMHg demethylation: (1) a reductive process, producing Hg0 and CH4 usually found in Hg-contaminated sites (Barkay et al., 2003), and (2) an oxidative process producing HgII and CO2 (Oremland et al., 1991; Martín-Díaz et al., 2008; Kronberg et al., 2018). The reductive process is mediated through an enzyme encoded by the mer-operon (organomercurial-lyase pathway) allowing microorganisms to be Hg-resistant. Oxidative demethylation possibly occurs via co-metabolism of MMHg and small organic compounds (Oremland et al., 1991), but the mechanism remains unknown. Recently, lab experiments using pure cultures have identified MMHg degradation by (micro)aerobic methanotrophs (e.g., CH4-consuming microbes) (Lu et al., 2017).
Because diet can affect the ecology of the gut microbiota (Singh et al., 2017), it is possible that nutrient intake from different diets can control the rate of MMHg degradation by affecting the gut microbiota community structure. Using an in vitro model, we manipulated the nutrient composition of fecal slurries prepared from two healthy human individuals and tracked MMHg production and degradation using stable Hg isotopes. Changes in microbial community structure were assessed using high throughput sequencing of 16S rRNA gene amplicon. Our hypothesis is that changes in the structure and function of the gut microbiota community induced by changes in dietary nutrient intake can affect the demethylation rate of MMHg.
Ethics application (H09-17-12) was reviewed and approved by the Research Ethics Board at the University of Ottawa. No information from participants was collected for this study.
Fecal collection and incubation setupFresh fecal samples (< 1-hr old) collected from two healthy human volunteers were used as a proxy for distal gut microbiota. The volunteers had no history of gastrointestinal disorders, have consumed no antibiotics in the past six months, or any probiotics in the form of pills or food products in the last month. Feces were collected from two individuals in plastic commode provided. All manipulations were conducted under a nitrogen atmosphere devoided of oxygen in a Bactron anaerobic chamber (ShelLab). The fecal slurry was prepared by diluting 1-part fecal sample, 9-part degassed PBS prepared at pH 7.0. Fecal slurries were mechanically shaken and mixed in 100 mL glass bottle, in a 1:10 ratio with the respective medium: balanced (basal medium) (Olano-Martin et al., 2000), protein-rich (basal medium + 20g.L-1 peptone) and carbohydrate-rich (basal medium + 10g.L-1 glucose). Bottles were spiked with stable isotope enriched MM198Hg and 199Hg isotopes are added to the mix to measure Hg methylation and demethylation using LC-ICP-MS. This ratio was chosen to reflect that of MMHg:Hg in the gut environment. Final concentrations were [MMHg]=10 ng.g-1 and [HgII] = 1 ng.g-1, and were lower than levels typically found in fish meal. All treatments were performed in biological triplicates. Serum bottles containing the samples were crimped to ensure anaerobic conditions and wrapped in aluminum foil to limit light exposure. Bottles were placed in a shaking incubator at 37°C. Bottles were sampled at t = 0, 12, 24, 36 and 48 hr for Hg transformation analysis and t = 0 and 48 hr for microbial analysis. Samples were stored in 15 mL falcon tubes kept at -20°C for microbial community analysis and in small 7 mL scintillation vial kept at -80°C for Hg isotope analysis.
Probiotic and transfer experimentSutterella parvirubra (DSMZ-19354) and Acidaminococcus intestini (DSMZ-21505) were commercially obtained from the DSMZ to test for their demethylation potential. Both strains were grown as per DSMZ cultivation conditions. Strains were tested for their methylation and demethylation potential in the balanced medium. In another series of experiments, Sutterella parvirubra or Acidaminococcus intestini were added to individual B fecal slurry to test for their potential to induce a demethylation phenotype. Finally, to test the effect of transferring the gut microbiota of individual A into individual B, fecal slurry preparation of individuals A and B was added in 1:1 (5 mL of individual A and 5 mL of individual B) and 1:2 (3.3 mL of individual A and 6.7 mL of individual B) ratio.
Hg analysesSamples were prepared according to the modified protocol outlined by Batista et al. (2011). All analysis and mercury isotope separation were performed on an Agilent LC-ICP-MS system. Chromatographic separation of isotope MM198Hg (demethylation tracer), 199Hg (methylation tracer), 201Hg (internal standard), and 202Hg (ambient) was performed using a mobile phase consisting of 0.05% v/v mercaptoethanol, 0.4% m/v L-cysteine and 0.06 M ammonium acetate on Agilent 1200 Infinity LC system consisting of a 1260 Isocratic pump and 1260 Autosampler. The LC system was connected to the Agilent 7700x ICP-MS via Peek tubing and equipped with a low flow Micro Mist Nebulizer and quartz, low-volume Scott-type double-pass spray chamber. All calculations for quantifying excess of MM199Hg and MM198Hg were made according to Hintelmann and Evans (1997). Additional information can be found in supporting methods.
Microbial community analysis using 16S rRNA gene amplicon metagenomicDNA was extracted using the PowerSoil® DNA Isolation Kit (Mo Bio Laboratories, Carlsbad, CA, USA). Sequencing was performed by Molecular Research LP (Shallowater, TX, USA), which amplified DNA using a two-step PCR targeting the V4 hypervariable region of the 16S rRNA gene using the primer 515f (forward: GTGYCAGCMGCCGCGGTAA) and 806r (reverse: GGACTACNVGGGTWTCTAAT). A negative control of our DNA extraction kit was performed using PBS 1X as a sample. We were not able to obtain an amplicon and no band was observed on a gel electrophoresis; samples were sent to the sequencing facility which was also unable to reliably generate amplicons for sequencing.
Amplicon sequence datasets were analyzed using the Quantitative Insights into Microbial Ecology (QIIME) pipeline (Caporaso et al., 2010a). The data was quality filtered using QIIME default parameters (quality score = 25, min length = 200, max length = 1000). Additional quality filtering and operational taxonomic unit (OTU) clustering were performed the UCHIME algorithm to identify chimera sequences for removal against the Gold databases. Remaining sequences were clustered into OTU (97% identity) using Closed reference OTU picking with UCLUST to form the representative dataset. Taxonomical classification of the representative dataset was performed using Ribosomal Database Project’s classifier against the SILVA database as a reference using the default settings (DeSantis et al., 2006). The representative sequences for each OTU were aligned using PyNAST (Caporaso et al., 2010b).
Statistical analysisAll analyses were performed in R v. 3.3.2 (R Core Team 2017). Beta diversity, weighted Unifrac distances, were used to perform the principal coordinate analyses (PCoA) using the package Vegan (http://www.cran.r-project.org/package = vegan). Data separation in the PCoA was tested using a permutational multivariate analysis of variance (PERMANOVA; function Adonis in the Vegan package). Hierarchical cluster analysis was performed using weight Unifrac distance dissimilarity matrix Two-way analyses of variance (ANOVA) with Tukey multiple comparisons were used to evaluate differences in Hg methylation and demethylation potential between medium and incubation time.
Using fecal slurry incubations, we conducted a series of experiments aiming to evaluate the potential for the gut microbiota to affect Hg speciation under different nutritional profiles for two healthy human adult individuals, referred to as individuals A and B. At 48 hr post-incubation, MM198Hg levels in individual A fecal slurries incubated in protein-rich medium were undetectable (Fig. 1A, Fig. S1; p < 0.001). In the balanced diet treatment, MM198Hg concentrations decreased by 52.5%. No significant decrease was observed in the carbohydrate-rich treatment (Fig. 1A). Strikingly, MMHg degradation was muted in individual B samples (Fig. 1B; Fig. S1). We did not observe significant production of MM199Hg in any of the treatments, supporting previous reports that the gut microbiota is not a hotspot for MMHg formation (Fig. S2). Control abiotic experiments (balanced, carbohydrate- and protein-rich treatments amended with stable isotope) did not show changes in Hg isotope concentrations (MM198Hg and 199Hg; Fig. S3). These results suggest that i) the ability to degrade MMHg is individual dependent and ii) that protein amendments stimulate pathways that lead to MMHg degradation. All experiments were conducted in darkness, ruling out the possibility of abiotic demethylation of MMHg via photodegradation (Seller et al., 1996).
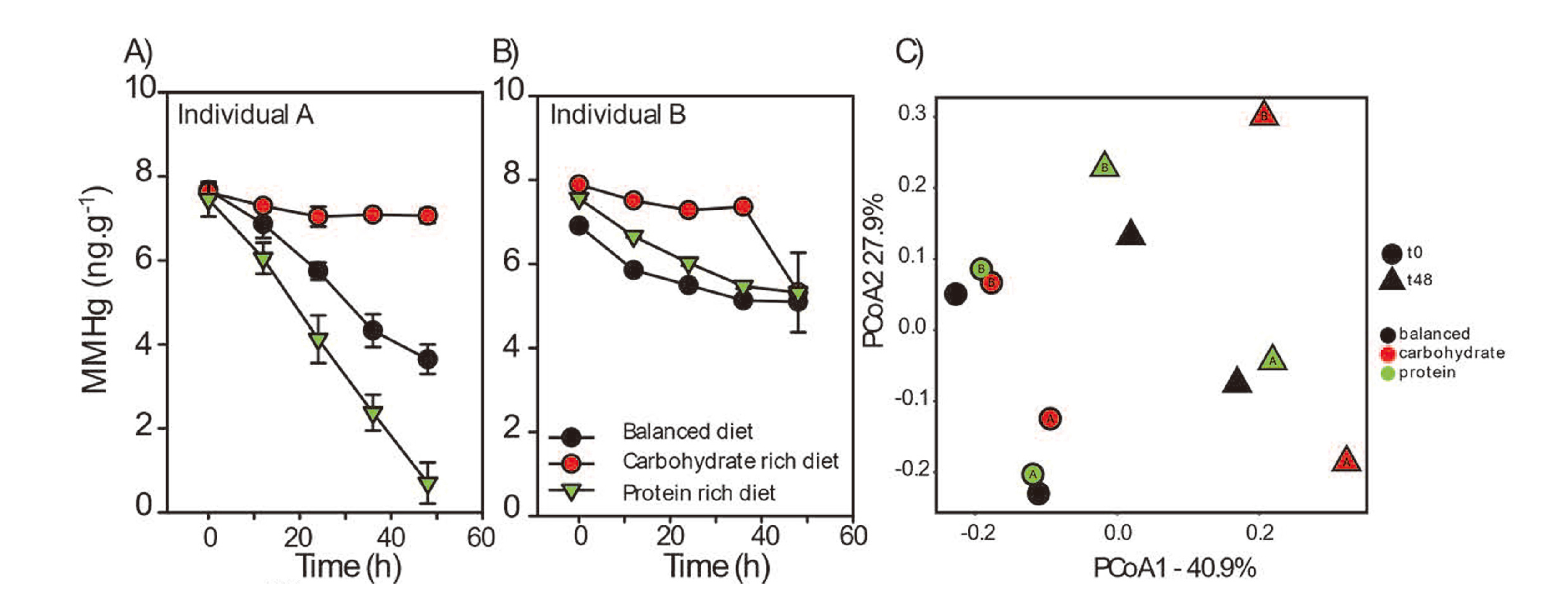
MM198Hg concentrations over time for individuals A (A) and B (B). Fecal slurries from individual A and B were prepared in balanced medium, balanced medium enriched with carbohydrates or balanced medium enriched with proteins. (C) A principal coordinates analysis based on characterization of the overall fecal community structure for individuals A and B. Weighted UniFrac metrics was calculated using QIIME to compute microbial β diversity. Sample dissimilarities were projected on a two-dimensional axis using principal coordinate analysis (PCoA). * indicates any significant difference between treatment and control.
Individuals A and B exhibited similar gut microbial community structures (Fig. 1C, Fig. 2), which were both dominated by Firmicutes and Bacteroidetes (> 80% in relative abundance), as expected from the gut microbiota of healthy human individuals (Ley et al., 2008). The addition of Hg did not significantly affect the microbial community structure suggesting that the Hg tracer added did not significantly alter the gut microbiota community structure (Fig. 2). Using PERMANOVA (Adonis), we are able to identify the incubation time (T0 vs T48) as a variable that significantly affected the microbial community structure in both individuals (Fig. 1C; p = 0.002). Carbohydrate-rich medium post-incubation also significantly shifted the microbial community structure that is different from the structure observed in the balanced and protein-rich medium (Fig. 1C; p = 0.007). The carbohydrate-rich treatment promoted the growth of Actinobacteria whereas balanced and protein-rich media saw an increased abundance of phyla that were already dominating the gut microbial profile at T0. The carbohydrate-rich treatment also decreased baseline MMHg degradation rates observed in individual A (Fig. S1). One possible explanation for the absence of MMHg degradation in the carbohydrate treatment could be the production of compounds inhibiting MMHg degradation. Indeed, Lu et al. (2017) showed that C1 compounds could inhibit oxidative MMHg degradation. Further investigation is required to determine if the same process takes place in the human gut environment.

(Top) Barplot showing relative abundance (phylum level) from the high-throughput 16S rRNA amplicon sequencing of the control experiment testing for the effect of the Hg tracer added. The table displays alpha diversity calculated for both individuals at the family level. An ANOVA did not detect any significant differences.
A more detailed analysis of the changes in the microbial community structure showed that individual A exhibited a significant increase in Sutterellaceae and Acidaminococcaceae (0.44 → 15.34%) in the protein-rich medium after 48 hr of treatment (Table 1) that was not observed for individual B (Table 1). Both bacterial families are known to be commensal of the human gut (D’Auria et al., 2011; Morotomi et al., 2011).
| Balanced (%) | Protein (%) | ||
|---|---|---|---|
| Individual A | Sutterellaceae | 0.08 → 0.33 | 0.22 → 7.87* |
| Acidaminococcaceae | 0.05 → 2.46* | 0.23 → 7.87* | |
| Individual B | Sutterellaceae | 0.08 → 0.04 | 0.07 → 0.04 |
| Acidaminococcaceae | N/A | N/A |
All values are relative proportion in percentage. (*) represent significant difference (p < 0.001). Acidaminococcaceae was not detected in individual B.
In a subsequent series of experiments, commercially obtained Sutterella parvirubra (DSMZ-19354) and Acidaminococcus intestini (DSMZ-21505) were tested for their MMHg degradation capabilities in balanced and protein-rich media. A. intestini incubated in protein-rich medium exhibited significantly higher demethylation rates than in the balanced medium. S. parvirubra did not show a significant change in MMHg degradation rates compare to control (Fig. 3). When both strains were combined together (1:1 ratio), MMHg demethylation rate was significantly higher than control but much lower than what was observed in the fecal slurry experiments (0.14 nM.g-1.hr-1 of MM198Hg, vs 6.7 nM.g-1.hr-1 of MM198Hg, respectively; Fig. 3).
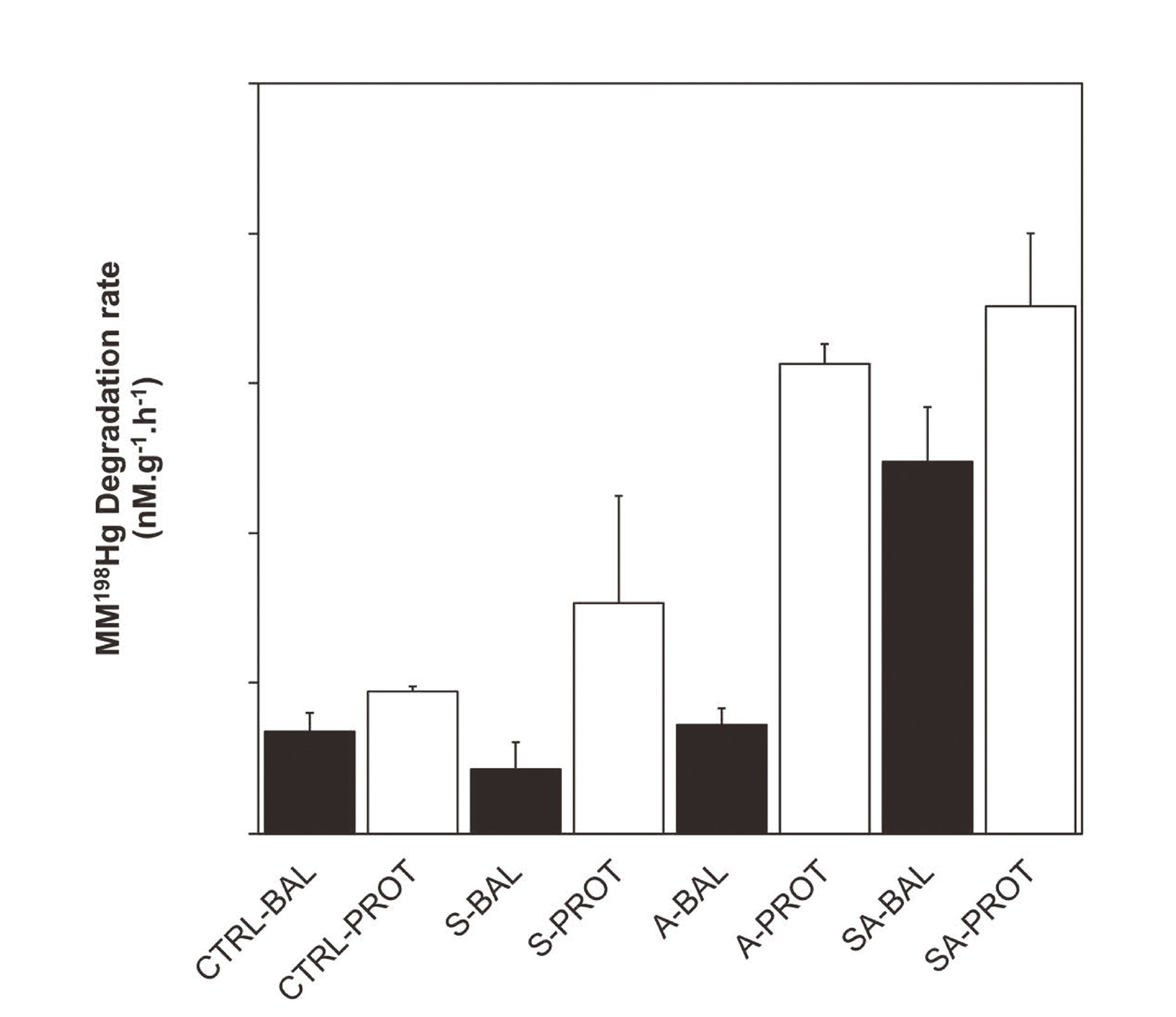
Demethylation from pure culture Sutterella parivubra (S) and Acidaminococcus intestini (A) in balanced (BAL) and protein-rich (PROT) media. Control treatments consisted of media with no microbial amendments. Letters represent significant differences: (a) indicate significant differences between nutrient amendment added while (b) indicate significant differences between treatment and abiotic control (ANOVA and Tukey post-hoc; p < 0.001).
The next experiment was to add S. parvirubra and A. intestini strains to fecal slurries collected from individual B to test whether they would enhance the demethylation of MMHg. We did not observe significant changes in MMHg metabolism with the addition of S. parvirubra or A. intestini in individual B fecal slurries (Fig. 4). These data suggest that MMHg demethylation does not result from the isolated activity of these two single strains. These results indicate that the increased abundance of these two strains in the demethylating individual likely reflects more complex changes within the gut microbial community. The enhanced degradation of MMHg as a result of protein addition could be explained by syntrophic interactions, in which microorganisms are linked via metabolic handoffs (Abreu and Taga, 2016; Anantharaman et al., 2016). Syntrophic interactions have already been identified as important in the production of MMHg (Yu et al., 2018) and deserve further investigations to evaluate their role in MMHg degradation.
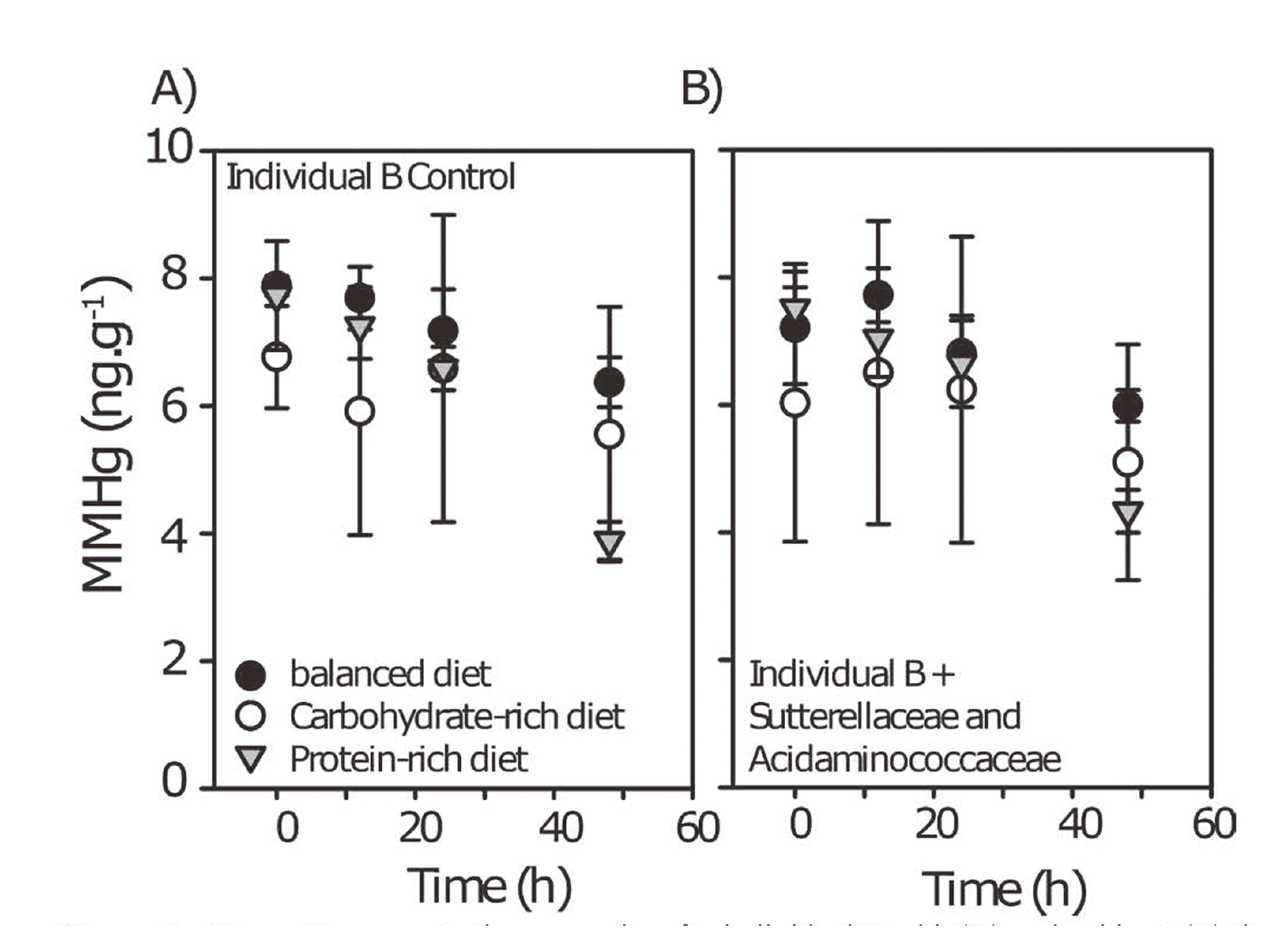
MM198Hg concentrations over time for individual B with (A) and, without (B) the addition of S. parivubra and A. intestini.
Finally, to test whether the absence of MMHg degradation in individual B was the result of a community-level inhibitory process, we performed a series of fecal mixing experiments. In these experiments, fecal samples from individual A (strong demethylator phenotype) and B (weak demethylator phenotype) were mixed in different ratio. We observed that degradation of MMHg was increasingly higher with increasing ratio of fecal samples from individual A (Fig. 5). These data suggest that the microbial community of individual B did not inhibit the degradation of MMHg and that individual A fecal samples consistently exhibit a microbial community that can be stimulated to enhance MMHg degradation in vitro. We tested for the presence of genes encoding for the mer operon to test if these genes were responsible for the higher demethylation capacity of the microbiota of individual A. None of the mer operon genes that we targeted were present in the gut microbiota of either individual A or B (data not shown).
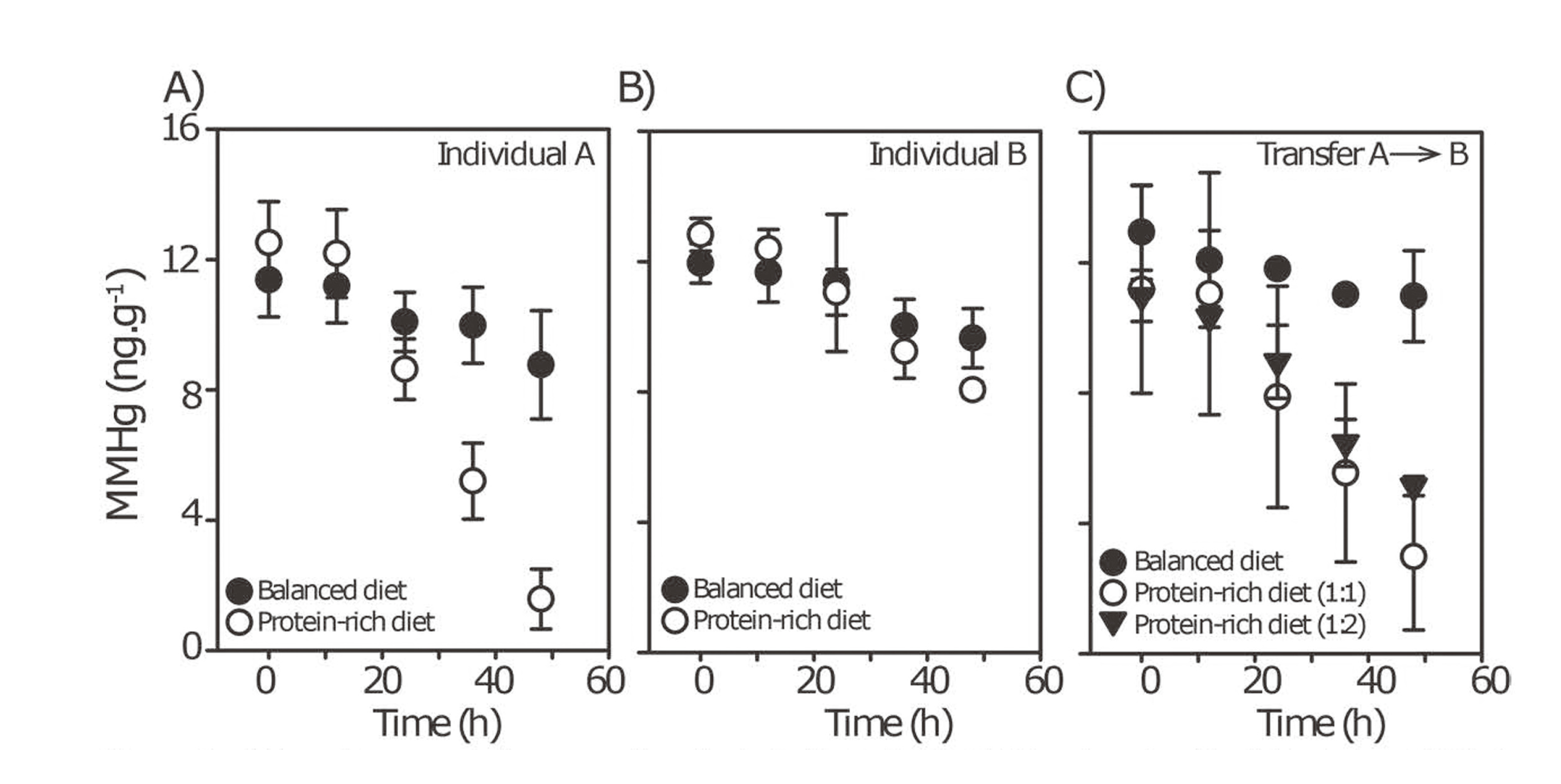
MM198Hg concentrations over time for individuals A (A), B (B) and a mix of individual A and B (1:1 and 1:2, A:B, respectively) (C) prepared in balanced medium and balanced medium amended with protein. Note that MM199Hg concentrations did not change across treatments suggesting the absence of methylation (Fig. S3). * indicates any significant difference between treatment and control.
We cannot yet offer a convincing mechanism explaining the quasi-complete demethylation phenotype observed for individual A in the presence of proteins. Nevertheless, these data are the first to show a nutrient dependency on the ability of the human gut microbiota to demethylate MMHg in vitro. We are currently gathering evidence testing how widespread this superdemethylation phenotype is at the human population level. This research contributes to the understanding of the roles that the gut microbiota and nutrient intake have on Hg metabolism. Should a reproducible mechanistic relationship between diet, microbiota community structure and function, and MMHg demethylation phenotype be identified, this research could lead to a shift in how risk assessment is conducted.
Supporting informationContains additional methodological details and additional results further supporting the main findings.
Our work was funded by NSERC Discovery (AJP, HMC), Accelerator grant (AJP), CFI funding (AJP, HMC), Canada Research Chair (HMC) and an NSERC CREATE-REACT scholarship to GG. We are grateful to the participants of this study that have contributed fecal samples and for Poulain and Chan lab members for stimulating discussion. We would like to thank Morgan McMillan for her help with the analyses.
Conflict of interestThe authors declare that there is no conflict of interest.