Abstract
Our aim was to demonstrate the significance of miRNA, lncRNA, and circRNA in the transformation of human bronchial epithelial cells induced by benzo(a)pyrene (BaP), and to investigate their regulatory networks. Hierarchical clustering, gene ontology (GO) analysis, Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis and network regulation analysis were used to analyze the high-throughput sequencing results of human bronchial epithelial cell line BEAS-2B and BaP-transformed BEAS-2B cells (BEAS-2B-T). 76,191,786 and 3,431differentially-expressed miRNA, lncRNA, mRNA and circRNA were detected, respectively; 43 miRNA, 48 lncRNA, 438 mRNA and 2,079 circRNA were up-regulated; 33 miRNA, 143 lncRNA, 348 mRNA and 1,352 circRNA were down-regulated. Through GO analysis, 257 biological process (BP) terms, 12 cell composition (CC) terms and 49 molecular function (MF) terms were found in differentially-expressed lncRNA; 143 BP terms, 32 CC terms, and 48 MF terms were found in differentially-expressed circRNA. Pathways of KEGG analysis of lncRNA and circRNA could be classified into the categories “human diseases” and “organism systems”. From miRNA-circRNA, circRNA-mRNA, and lncRNA-circRNA networks analysis, we found that mir-137, circ-RPS5, circ-ZNF292, circ-ERBB2IP, circ-SEMA3C, circ-IGF1R, circ-RTN4, APOC1, and CDKN2A may be of great significance for cell transformation. From the analysis of miRNA, lncRNA, mRNA, and circRNA networks, we found that PDGFRB, lncRNA RGMB-AS1, circ-ZNF292 are associated with miR-138-5p. Our study shows that miRNA, lncRNA, and circRNA have a significant regulatory role in the transformation of human bronchial epithelial cell induced by BaP.
INTRODUCTION
Lung cancer is the leading cause of cancer-related death worldwide. Several studies have shown that the occurrence of lung cancer is closely related to environmental carcinogens (Torre et al., 2016). Benzo(a)pyrene (BaP) is a chemical environmental carcinogen; it is a polycyclic aromatic compound composed of a benzene ring and a pyrene molecule. BaP is an important chemical carcinogen in tobacco (Pfeifer et al., 2002). BaP has been widely used to study the molecular mechanisms of lung cancer (Yeo et al., 2014; Hung et al., 2015). The mechanisms promoting lung cancer are still unclear, and its early diagnosis and treatment are still unsatisfactory. Therefore, it is necessary to ascertain the important regulatory mechanism of carcinogenesis and to find early biomarkers.
Recent studies have found that noncoding RNA, microRNA (miRNA) and long noncoding RNA (lncRNA) have regulatory significance in tumors (Huang et al., 2013; Qu et al., 2015). Another kind of noncoding circular RNA (circRNA) was first discovered in 1980. CircRNA has a closed loop structure and usually consists of more than one exon (Jeck et al., 2013). In 2012, researchers confirmed that circRNA is a predominant transcript isoform in human genes (Salzman et al., 2012). In 2013, Nature published two articles in the same issue about the circRNA functions (Memczak et al., 2013; Hansen et al., 2013). One of these articles showed that circ-CDR1is in up-regulated in the nerve tissue of mice and humans. The circ-CDR1 contains several miR-7 binding sites and acts as a competing endogenous RNA (ceRNA). circ-CDR1 overexpression in zebrafish embryos leads to a decrease in midbrain volume, consistent with the effect of silencing miR-7. The “miRNA sponge” function of circRNA has been reviewed in many journals, including Science (Wilusz and Sharp, 2013; Taulli et al., 2013; Hentze and Preiss, 2013). Although research on circRNA is still in its early stages, some studies have suggested a close relationship between circRNA and cancer (Hsiao et al., 2017; Yao et al., 2017). It is also worth mentioning that there were two articles published in March 2017, showing that circRNA can regulate protein translation (Legnini et al., 2017; Pamudurti et al., 2017).
In this paper, through high-throughput sequencing, we analyzed differentially-expressed miRNA, lncRNA, circRNA, and mRNA of immortalized human bronchial epithelial cell line BEAS-2B, and of BaP-transformed BEAS-2B cells (BEAS-2B-T). Gene ontology (GO) analysis, Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis and network regulation analysis were done based on the results of high-throughput sequencing.
MATERIALS AND METHODS
Cell culture
The immortalized human bronchial epithelial cell line BEAS-2B, and BaP-transformed BEAS-2B cells (BEAS-2B-T) were provided by the State Key Laboratory of Respiratory Disease, Institute for Chemical Carcinogenesis, Guangzhou Medical University, Guangzhou, China (Gao et al., 2013). BEAS-2B cells and BEAS-2B-T cells were cultured in RPMI-1640 medium containing 10% heat-inactivated fetal bovine serum and 1% penicillin-streptomycin (Invitrogen; Carlsbad, CA, USA). Cells were incubated at 37°C and 5% CO2. BEAS-2B cells and BEAS-2B-T cells were divided into two dishes when cell confluence reached 80%-90%. BEAS-2B and BEAS-2B-T cells were passaged once after 2 or 3 days. Both cell lines used for high-throughput sequencing were the third-generation cells after resuscitation. Approximately 1.0 × 106 BEAS-2B or BEAS-2B-T cells were inoculated into 10 cm dishes at the third-generation inoculation.
High-throughput sequencing analysis
Cell ribosomal RNA was removed by Ribo-Zero™rRNA Removal Kit (Epicentre, Madison, WI, USA). The processing of circRNA requires removal of linear RNA in the sample by RNaseR. Synthesis of double-stranded cDNA was performed by reverse transcription and then cDNA was end-repaired. After PCR amplification and purification, and library screening by quality test, the samples were finally sequenced by using the Illumina HiSeqTM 2500 sequencing platform (Illumina, San Diego, CA, USA). Bioinformatics analysis of the raw sequencing data was then carried out. Differentially-expressed lncRNAs were sought in the NCBI database (http://www.ncbi.nlm.nih.gov/) to determine their genome loci. We used ANNOVAR to compare the location of the ends of the circRNA splice site in the genome. The functional components of the circRNA were annotated by comparing multiple databases.
Differential expression analysis
Differentially-expressed miRNA, mRNA, lncRNA and circRNA were screened by fold change > 2.0 and p value < 0.05. Hierarchical clustering analysis (HCL) was used to determine the expression pattern of different transcripts under different experimental conditions. The results are presented using Tree view software (V.1.5). lncRNA and circRNA differential expression was further analyzed with biological pathway analysis, based on the KEGG database (http://www.genome.jp/), and by GO analysis.
miRNA, lncRNA, circRNA, and mRNA network analysis
Differentially-expressed miRNA, mRNA, lncRNA, and circRNA were identified from the database. Pearson’s correlation coefficient was used to measure the strength of the linear dependence of two variables. Pearson’s correlation coefficient value was calculated for the miRNA-circRNA, lncRNA-circRNA and circRNA-mRNA pairs. Pairs’ absolute coefficient value over 0.8 and P value below 0.05 were screened. The miRNA-circRNA, lncRNA-circRNA and circRNA-mRNA network maps were done by Cytoscape software (V.3.5.1). Differentially-expressed miRNA, lncRNA, circRNA, and mRNA were analyzed by Ingenuity Pathway Analysis (IPA) for overall regulatory network analysis based on Ingenuity Knowledge Base.
Statistical analysis
All experiments were performed using at least three biological replicates. The differences between groups were analyzed by independent sample t tests, while the differences between multiple groups were compared with ANOVA analysis. P values < 0.05 indicated that differences were statistically significant.
RESULTS
Expression profiles of differentially-expressed miRNA, lncRNA, mRNA, and circRNA
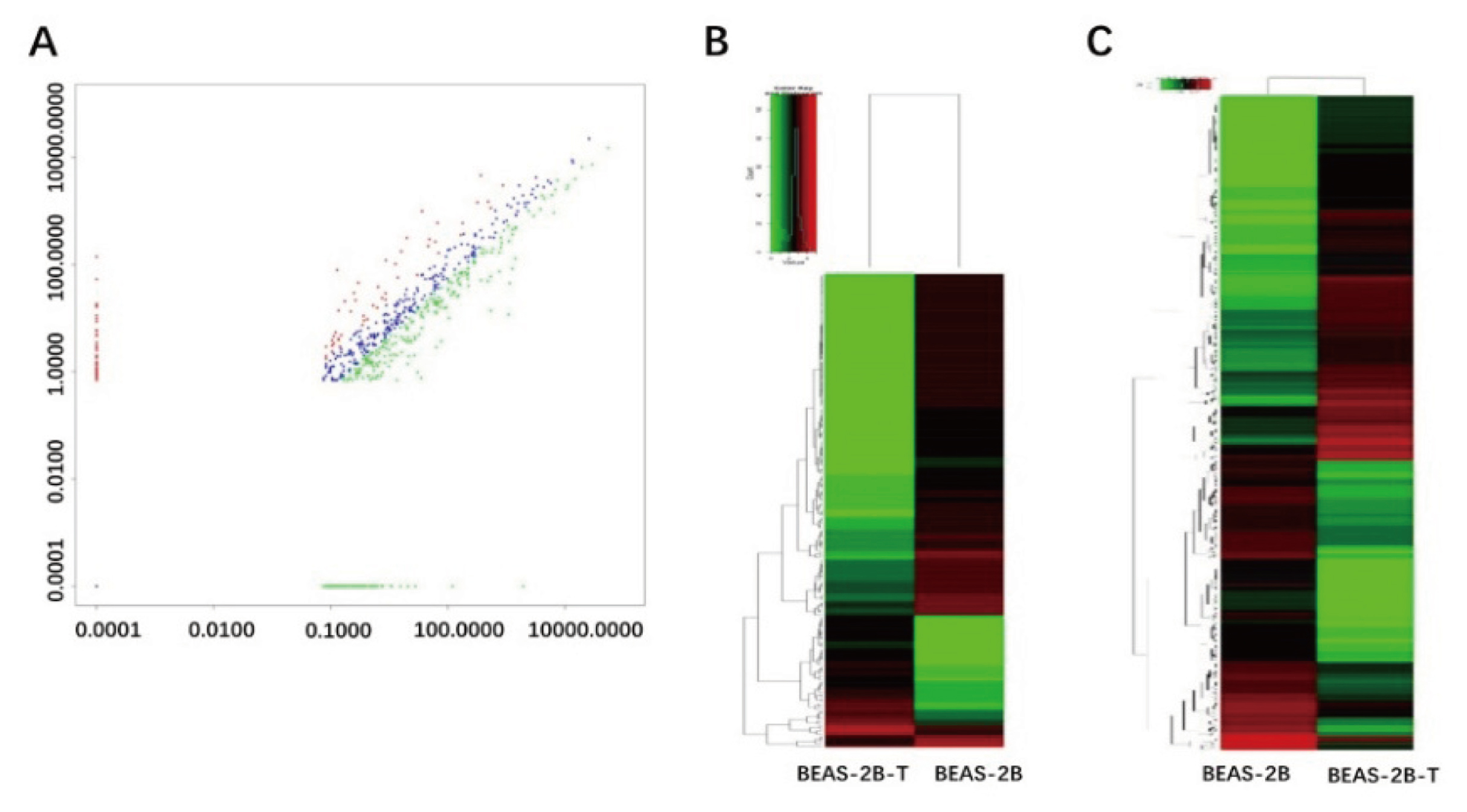
We obtained the total miRNA, mRNA, lncRNA and circRNA of immortalized human bronchial epithelial cell line BEAS-2B and the BaP-transformed cell line BEAS-2B-T through high-throughput sequencing. The scatter plots of miRNA show the difference of expression between two groups of miRNA (fold change > 2.0) (Fig. 1A). Seventy-six differentially-expressed miRNA were detected (fold change > 2.0, p value < 0.05). After BaP-induced malignant transformation 43 miRNA were up-regulated and 33 miRNA were down-regulated, relative to bronchial epithelial cells. Fold change > 2.0 and p value < 0.05 were used as screening criteria, 191 differentially-expressed lncRNA were detected by HCL, 48 of which were up-regulated and 143 down-regulated (Fig. 1B). Similarly, 786 differentially-expressed mRNA were detected by HCL, 438 of which were up-regulated and 348 down-regulated (Fig. 1C). Subgroup analysis showed genomic classification and distribution of total circRNA in both BEAS-2B and BEAS-2B-T cells (Fig. 2A, 2B). The results showed that exonic circRNA, intronic circRNA and intergenic circRNA accounted for 84%, 7% and 2% of total RNA in BEAS-2B cells, respectively. Compared with BEAS-2B cells, the distribution of exonic circRNA and intronic circRNA in BEAS-2B-T cells varied by 1%, but the rest remained unchanged. A total of 3,431 differentially-expressed circRNA were detected by HCL, of which 2,079 were up-regulated and 1,352 were down-regulated (fold change > 2.0, p value < 0.05). Interestingly, from the heat map we observe that the total expression of circRNA in BEAS-2B-T cells was much higher than in BEAS-2B cells (Fig. 2C).
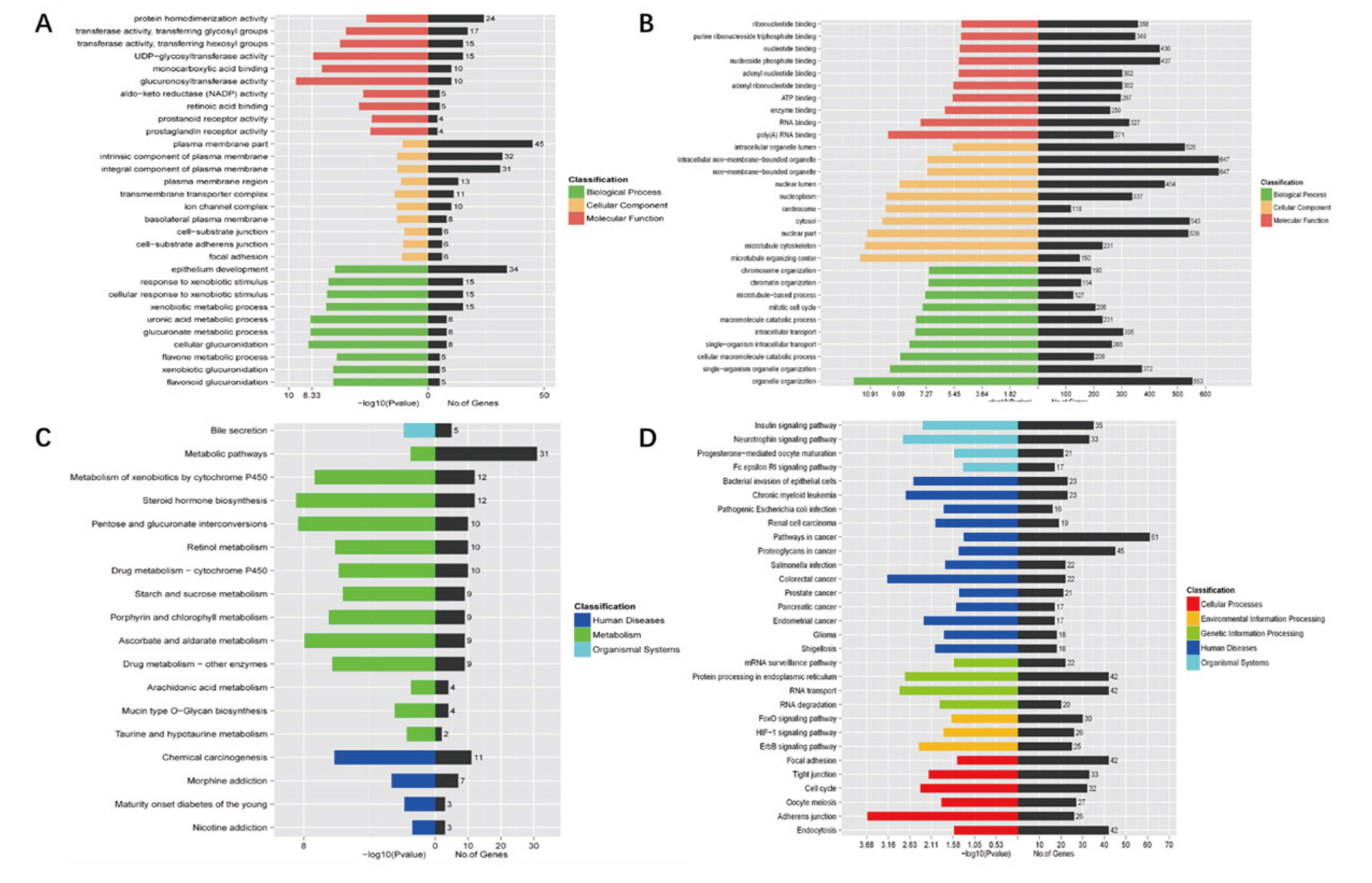
In the GO analysis, prediction terms with p value < 0.05 were selected. The GO analysis was divided into three parts: biological process (BP), cell composition (CC) and molecular function (MF). A total of 257 BP terms, 12 CC terms, and 49 MF terms were found in differentially-expressed lncRNA. A total of 143 BP terms, 32 CC terms, and 48 MF terms were found in circRNA differentially-expressed genes. We have listed three parts of the top 10 generally changed GO terms, which are related to the research background (Fig. 3A, 3B). We found that the lncRNA differentially-expressed genes, partially meaningful BP terms, CC terms, and MF terms were related to transferase activity, plasma membrane, and epithelial development. Moreover, the pathways of KEGG pathway analysis of lncRNA differentially-expressed genes were divided into human diseases, metabolism, and organism systems (Fig. 3C). The circRNA differentially-expressed genes, partially meaningful BP terms, CC terms, and MF terms are associated with nucleotide binding, organelles, and intracellular transport. We have listed the top 30 pathways of KEGG pathway analysis of circRNA differentially-expressed genes; these pathways could be divided into cellular processes, environmental information processing, genetic information processing, human diseases, and organism systems (Fig. 3D).
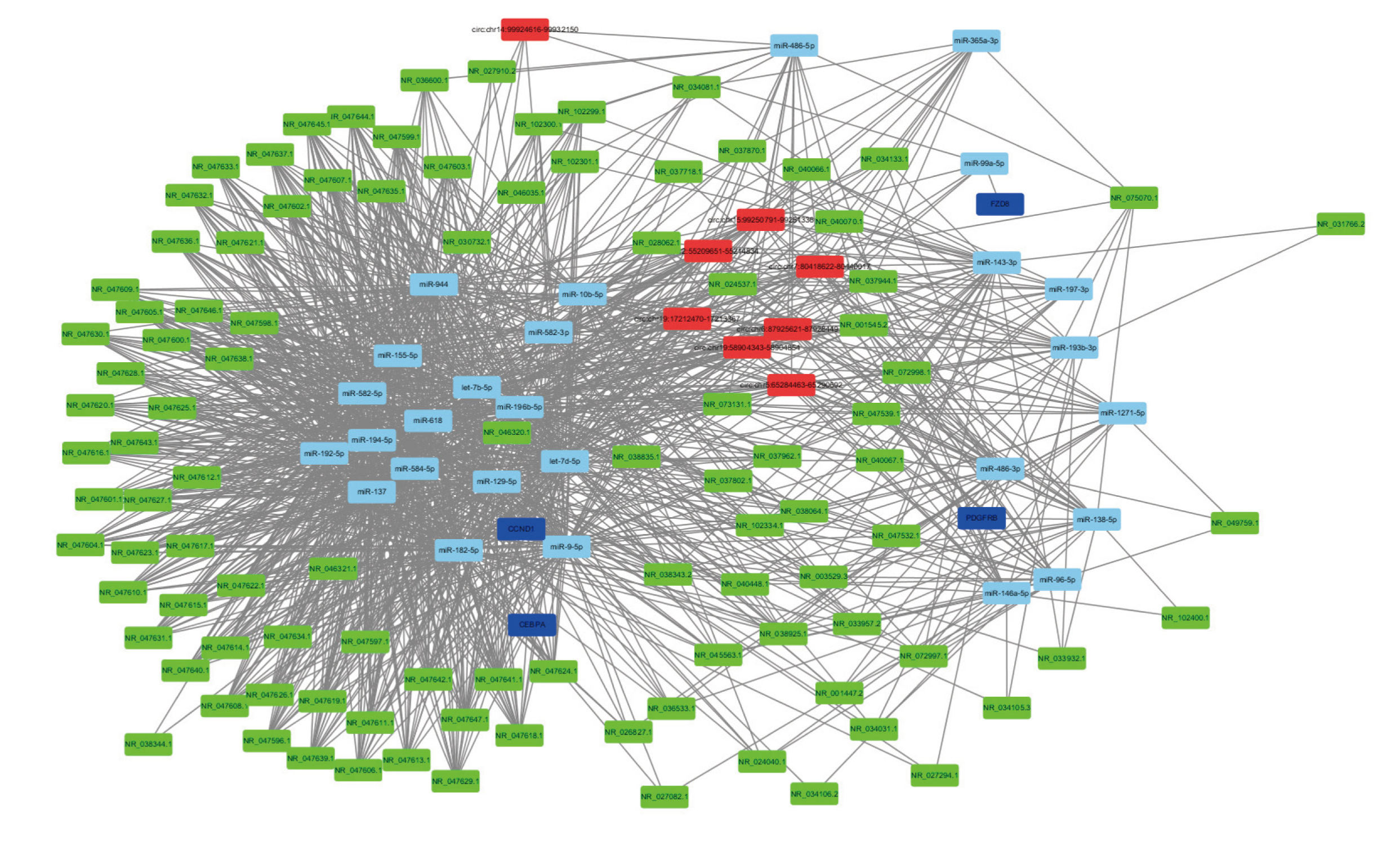
miRNA-circRNA, circRNA-mRNA, and lncRNA-circRNA network analysis was based on differentially-expressed miRNA, circRNA, lncRNA and mRNA (fold change > 2.0, p value < 0.05). Sub-networks with the highest number of interactions are presented in detail. In the miRNA-circRNA network, blue represents miRNA, and red represents circRNA (Fig. 4A). From the network, we found that mir-137 was associated with circ-RPS5 (has_circ:chr19:58904343-58904854), circ-ZNF292 (hsa_circ:chr6:87925621-87928449), circ-ERBB2IP (hsa_circ:chr5:65284463-65290692), circ-SEMA3C (hsa_circ:chr7:80418622-80440017), circ-IGF1R (hsa_circ:chr15:99250791-99251336), and circ-RTN4 (hsa_circ:chr2:55209651-55214834). The important role of mir-137 in lung cancer has been widely reported. There are articles showing that up-regulation of mirRNA-137 in non-small cell cancer inhibits TFAP2C, thus promoting tumor invasion and metastasis (Chang et al., 2017). In the circRNA-mRNA network, blue represents mRNA, and green represents circRNA (Fig. 4B). APOC1 (NM_001645), CDKN2A (NM_058195) and many other mRNA present in the network are important in tumor occurrence (Sun et al., 2016; Spitzwieser et al., 2017). Some of mRNA are related to two or even three circRNA. circRNA has the potential of extensively regulating the process of cell transformation. In the lncRNA-circRNA network, blue represents lncRNA, and red represents circRNA (Fig. 4C), three lncRNA, three circRNA, and four relationships were obtained.

Differentially-expressed miRNA, lncRNA, circRNA, and mRNA were analyzed by IPA for overall regulatory network analysis. Sub-networks with a high number of interactions are presented in detail in Fig. 5. In the network, navy blue represents mRNA, light blue represents miRNA, green represents lncRNA, and red represents circRNA. From numerous networks of mRNA-miRNA-lncRNA-circRNA, we found a sub-network with potentially important regulatory significance. PDGFRB, lncRNA RGMB-AS1 (NR_033932.1) and circ-ZNF292 (hsa_circ:chr6:87925621-87928449) are all associated with miR-138-5p. Many studies suggest that mir-138-5p plays an important role in cancer (Yang et al., 2017; Yu et al., 2018). miR-138-5p can reverse gefitinib resistance in non-small cell lung cancer cells (Gao et al., 2014). lncRNA RGMB-AS1 down-regulation significantly inhibited the growth of adenoma of lung cancer (Li et al., 2016).
DISCUSSION
Although lung cancer has been studied for decades, the mechanism is still unclear. Lung cancer clinical diagnosis and treatment are still not optimal. Lung cancer survival rate is only 11% (Verdecchia et al., 2007). Recent studies have found that ncRNA plays an important role in epigenetic inheritance and is closely related to tumor occurrence (Knowling and Morris, 2011). In recent years, the functional significance of miRNA, lncRNA, and circRNA have been reported. miRNA is a family of small ncRNAs that negatively regulate gene expression by interacting with the 3’UTR of target mRNA (Crea et al., 2014). Many studies have been published on the relationship between miRNA and lung cancer. miRNA-605 promotes cell proliferation, migration and invasion in non-small cell lung cancer (NSCLC) by targeting LATS2 (Ye et al., 2017). A meta-analysis of miRNA and NSCLC was done based on PUBMED, EMBASE, Web of Science and included data of 12 studies. This analysis suggested that patients with higher expression of miRNA tended to show lower survival rate, hazard ratio (HR) 2.49, 95% confidence interval (Cl): 1.84-3.37, and showed that miRNA can be used as potential prognostic markers of NSCLC (Xu et al., 2013). lncRNA and circRNA’s biological function mechanism is more inclined to treat them as upstream regulatory substances to regulate miRNA expression (Song et al., 2014; Kulcheski et al., 2016). Some papers indicate that some circRNA can be directly translated into protein (Legnini et al., 2017; Pamudurti et al., 2017).
In our experiment, we compared bronchial epithelial cells with BaP-transformed bronchial epithelial cells; 76, 786, 191 and 3,431 differentially-expressed miRNA, mRNA, lncRNA and circRNA were detected, respectively (fold change > 2.0, p value < 0.05). We found that the expression of circRNA in BEAS-2B cells was very low. However, the expression of numerous circRNA (such as circ-RMRP, circ-HIST1H2AB and circ-RPS5) was extremely high in BEAS-2B-T cells. A total of 2,079 circRNA were up-regulated and 1,352 were down-regulated in BEAS-2B-T cells. We thus speculate that circRNA plays a significant role in the malignant transformation of bronchial epithelial cells induced by BaP. A report shows that the overexpression of ALDH1A1 in BEAS-2B cells increased the expression of stem cell markers, facilitated cell transformation, promoted migratory ability, and enhanced drug resistance (Liu et al., 2016). In our study, we found that ALDH1A1 expression in BEAS-2B-T is significantly higher than in BEAS-2B cells. This further showed that ALDH1A1 may play an important role in malignant transformation of bronchial epithelial cells. In another report, lncRNA-DQ786227 was overexpressed; it was a functional ncRNA in BEAS-2B cell malignant transformation induced by benzo(a)pyrene (Gao et al., 2013). In contrast, we did not detect significant overexpression of lncRNA-DQ786227 in our high-throughput sequencing analysis. We speculate that differences in results are due to the different technologies used. We used high-throughput sequencing, while other studies used quantitative PCR. In our future studies, we will use more accurate methods to detect the expression of this lncRNA and analyze its related network.
Regulatory network analysis has been widely used in ncRNA research. lncRNA-mRNA and circRNA-miRNA co-expression network analysis was performed based on microarrays during osteoclastogenesis (Dou et al., 2016). In our study, lung cancer-related regulatory network, combining miRNA, mRNA, lncRNA, and circRNA by IPA was performed for the first time. From our network analysis, we conclude that PDGFRB, lncRNA-RGMB-AS1, circ-ZNF292, and miR-138-5p may be an important regulatory network in BaP-induced malignant bronchial epithelial cells. In a previous report, it was shown that GPR124, a target molecule of mir-138-5p, was important for resistance to gefitinib (Gao et al., 2014). In our study, we did not find significantly different expression of GPR124 after cell transformation in BEAS-2B-T cells. This shows that different miRNA targets may have different biological effects.
In conclusion, differently expressed miRNA, lncRNA, circRNA, and mRNA of BEAS-2B cells and BEAS-2B-T cells were detected by high-throughput sequencing. And we analyzed the result by using GO analysis, KEGG pathway analysis and network regulation analysis, to explore the association between miRNA, lncRNA, and circRNA and lung cancer.
ACKNOWLEDGMENTS
This project was supported by the National Natural Science Foundation of China (Grant No:81273116), the Guangdong Provincial Natural Science Foundation (S2013010015153), the Science Foundation of Guangdong Medical University (M2013004), and the National College Students’ Innovation and Entrepreneurship Training Program (201610571008).
Conflict of interest
The authors declare that there is no conflict of interest.
REFERENCES
- Crea, F., Clermont, P.L., Parolia, A., Wang, Y. and Helgason, C.D. (2014): The non-coding transcriptome as a dynamic regulator of cancer metastasis. Cancer Metastasis Rev., 33, 1-16.
- Chang, T.H., Tsai, M.F., Gow, C.H., Wu, S.G., Liu, Y.N., Chang, Y.L., Yu, S.L., Tsai, H.C., Lin, S.W., Chen, Y.W., Kuo, P.Y., Yang, P.C. and Shih, J.Y. (2017): Upregulation of microRNA-137 expression by Slug promotes tumor invasion and metastasis of non-small cell lung cancer cells through suppression of TFAP2C. Cancer Lett., 402, 190-202.
- Dou, C., Cao, Z., Yang, B., Ding, N., Hou, T., Luo, F., Kang, F., Li, J., Yang, X., Jiang, H., Xiang, J., Quan, H., Xu, J. and Dong, S. (2016): Changing expression profiles of lncRNAs, mRNAs, circRNAs and miRNAs during osteoclastogenesis. Sci. Rep., 6, 21499.
- Gao, L., Mai, A., Li, X., Lai, Y., Zheng, J., Yang, Q., Wu, J., Nan, A., Ye, S. and Jiang, Y. (2013): LncRNA-DQ786227-mediated cell malignant transformation induced by benzo(a)pyrene. Toxicol. Lett., 223, 205-210.
- Gao, Y., Fan, X., Li, W., Ping, W., Deng, Y. and Fu, X. (2014): miR-138-5p reverses gefitinib resistance in non-small cell lung cancer cells via negatively regulating G protein-coupled receptor 124. Biochem. Biophys. Res. Commun., 446, 179-186.
- Huang, T., Alvarez, A., Hu, B. and Cheng, S.Y. (2013): Noncoding RNAs in cancer and cancer stem cells. Chin. J. Cancer, 32, 582-593.
- Hansen, T.B., Jensen, T.I., Clausen, B.H., Bramsen, J.B., Finsen, B., Damgaard, C.K. and Kjems, J. (2013): Natural RNA circles function as efficient microRNA sponges. Nature, 495, 384-388.
- Hentze, M.W. and Preiss, T. (2013): Circular RNAs: splicing’s enigma variations. EMBO J., 32, 923-925.
- Hung, J.Y., Chang, W.A., Tsai, Y.M., Hsu, Y.L., Chiang, H.H., Chou, S.H., Huang, M.S. and Kuo, P.L. (2015): Tricetin, a dietary flavonoid, suppresses benzo(a)pyrene-induced human non-small cell lung cancer bone metastasis. Int. J. Oncol., 46, 1985-1993.
- Hsiao, K.Y., Lin, Y.C., Gupta, S.K., Chang, N., Yen, L., Sun, H.S. and Tsai, S.J. (2017): Noncoding effects of circular RNA CCDC66 promote colon cancer growth and metastasis. Cancer Res., 77, 2339-2350.
- Jeck, W.R., Sorrentino, J.A., Wang, K., Slevin, M.K., Burd, C.E., Liu, J., Marzluff, W.F. and Sharpless, N.E. (2013): Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA, 19, 141-157.
- Knowling, S. and Morris, K.V. (2011): Non-coding RNA and antisense RNA. Nature’s trash or treasure? Biochimie, 93, 1922-1927.
- Kulcheski, F.R., Christoff, A.P. and Margis, R. (2016): Circular RNAs are miRNA sponges and can be used as a new class of biomarker. J. Biotechnol., 238, 42-51.
- Li, P., Zhang, G., Li, J., Yang, R., Chen, S., Wu, S., Zhang, F., Bai, Y., Zhao, H., Wang, Y., Dun, S., Chen, X., Sun, Q. and Zhao, G. (2016): Long noncoding RNA RGMB-AS1 indicates a poor prognosis and modulates cell proliferation, migration and invasion in lung adenocarcinoma. PLoS One, 11, e0150790.
- Liu, Y., Lu, R., Gu, J., Chen, Y., Zhang, X., Zhang, L., Wu, H., Hua, W. and Zeng, J. (2016): Aldehyde dehydrogenase 1A1 up-regulates stem cell markers in benzo[a]pyrene-induced malignant transformation of BEAS-2B cells. Environ. Toxicol. Pharmacol., 45, 241-250.
- Legnini, I., Di Timoteo, G., Rossi, F., Morlando, M., Briganti, F., Sthandier, O., Fatica, A., Santini, T., Andronache, A., Wade, M., Laneve, P., Rajewsky, N. and Bozzoni, I. (2017): Circ-ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol. Cell, 66, 22-37.e9.
- Memczak, S., Jens, M., Elefsinioti, A., Torti, F., Krueger, J., Rybak, A., Maier, L., Mackowiak, S.D., Gregersen, L.H., Munschauer, M., Loewer, A., Ziebold, U., Landthaler, M., Kocks, C., le Noble, F. and Rajewsky, N. (2013): Circular RNAs are a large class of animal RNAs with regulatory potency. Nature, 495, 333-338.
- Pfeifer, G.P., Denissenko, M.F., Olivier, M., Tretyakova, N., Hecht, S.S. and Hainaut, P. (2002): Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene, 21, 7435-7451.
- Pamudurti, N.R., Bartok, O., Jens, M., Ashwal-Fluss, R., Stottmeister, C., Ruhe, L., Hanan, M., Wyler, E., Perez-Hernandez, D., Ramberger, E., Shenzis, S., Samson, M., Dittmar, G., Landthaler, M., Chekulaeva, M., Rajewsky, N. and Kadener, S. (2017): Translation of CircRNAs. Mol. Cell, 66, 9-21.
- Qu, S., Yang, X., Li, X., Wang, J., Gao, Y., Shang, R., Sun, W., Dou, K. and Li, H. (2015): Circular RNA: A new star of noncoding RNAs. Cancer Lett., 365, 141-148.
- Salzman, J., Gawad, C., Wang, P.L., Lacayo, N. and Brown, P.O. (2012): Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One, 7, e30733.
- Song, X., Cao, G., Jing, L., Lin, S., Wang, X., Zhang, J., Wang, M., Liu, W. and Lv, C. (2014): Analysing the relationship between lncRNA and protein-coding gene and the role of lncRNA as ceRNA in pulmonary fibrosis. J. Cell. Mol. Med., 18, 991-1003.
- Sun, Y., Zhang, J., Guo, F., Zhao, W., Zhan, Y., Liu, C., Fan, Y. and Wang, J. (2016): Identification of Apolipoprotein C-I Peptides as a potential biomarker and its biological roles in breast cancer. Med. Sci. Monit., 22, 1152-1160.
- Spitzwieser, M., Entfellner, E., Werner, B., Pulverer, W., Pfeiler, G., Hacker, S. and Cichna-Markl, M. (2017): Hypermethylation of CDKN2A exon 2 in tumor, tumor-adjacent and tumor-distant tissues from breast cancer patients. BMC Cancer, 17, 260.
- Taulli, R., Loretelli, C. and Pandolfi, P.P. (2013): From pseudo-ceRNAs to circ-ceRNAs: a tale of cross-talk and competition. Nat. Struct. Mol. Biol., 20, 541-543.
- Torre, L.A., Siegel, R.L. and Jemal, A. (2016): Lung cancer statistics. Adv. Exp. Med. Biol., 893, 1-19.
- Verdecchia, A., Francisci, S., Brenner, H., Gatta, G., Micheli, A., Mangone, L. and Kunkler, I.; EUROCARE-4 Working Group. (2007): Recent cancer survival in Europe: a 2000-02 period analysis of EUROCARE-4 data. Lancet Oncol., 8, 784-796.
- Wilusz, J.E. and Sharp, P.A. (2013): Molecular biology. A circuitous route to noncoding RNA. Science, 340, 440-441.
- Xu, T.P., Zhu, C.H., Zhang, J., Xia, R., Wu, F.L., Han, L., Shen, H., Liu, L.X. and Shu, Y.Q. (2013): MicroRNA-155 expression has prognostic value in patients with non-small cell lung cancer and digestive system carcinomas. Asian Pac. J. Cancer Prev., 14, 7085-7090.
- Yeo, C.D., Kim, J.W., Ha, J.H., Kim, S.J., Lee, S.H., Kim, I.K. and Kim, Y.K. (2014): Chemopreventive effect of phosphodieasterase-4 inhibition in benzo(a)pyrene-induced murine lung cancer model. Exp. Lung Res., 40, 500-506.
- Yang, Q., Wang, X., Tang, C., Chen, X. and He, J. (2017): H19 promotes the migration and invasion of colon cancer by sponging miR-138 to upregulate the expression of HMGA1. Int. J. Oncol., 50, 1801-1809.
- Yao, J.T., Zhao, S.H., Liu, Q.P., Lv, M.Q., Zhou, D.X., Liao, Z.J. and Nan, K.J. (2017): Over-expression of CircRNA_100876 in non-small cell lung cancer and its prognostic value. Pathol. Res. Pract., 213, 453-456.
- Ye, Y., Zhuang, J., Wang, G., He, S., Ni, J., Xia, W. and Wang, J. (2017): microRNA-605 promotes cell proliferation, migration and invasion in non-small cell lung cancer by directly targeting LATS2. Exp. Ther. Med., 14, 867-873.
- Yu, Z., Wang, Z., Li, F., Yang, J. and Tang, L. (2018): miR-138 modulates prostate cancer cell invasion and migration via Wnt/β-catenin pathway. Mol. Med. Rep., 17, 3140-3145.