Letter
Disulfiram facilitates ataxin-3 nuclear translocation and potentiates the cytotoxicity in a cell model of SCA3
2019 Volume 44 Issue 8 Pages 535-542
Details
2019 Volume 44 Issue 8 Pages 535-542
Spinocerebellar ataxia type 3 (SCA3) is caused by the expansion of a glutamine-encoding CAG repeat in the ATXN3 gene encoding the protein ataxin-3. The nuclear presence of polyglutamine-expanded ataxin-3 is of critical importance for the pathogenesis of SCA3. Disulfiram, an FDA-approved drug for alcoholism, has also garnered attention in cancer treatment. However, it has shown toxicity in the nervous system. Bearing this in mind, we treated cells expressing ataxin-3 with disulfiram to measure several pathogenic cascades of SCA3, including aggregate formation, soluble ataxin-3 expression and nuclear localization of ataxin-3 and the cytotoxicity, which assess the direct effect of disulfiram on SCA3 cell models. To our knowledge, this is direct evidence that disulfiram elevated the nuclear localization of polyglutamine-expanded ataxin-3 and enhanced the cytotoxicity in a cell model of SCA3. Furthermore, disulfiram did not affect the aggregate formation of polyglutamine-expanded ataxin-3 at least at a single dose. Our findings repurpose disulfiram as a modulator of ataxin-3 nuclear transport that aggravates the pathology of SCA3, which is a new target for disulfiram. This study also represents an important example of determining novel side effects in pre-existing drugs. This study suggests that caution may be warranted when this compound is used to treat alcohol abuse or cancer in patients carrying a SCA3-causing mutation.
Spinocerebellar ataxia type 3 (SCA3), also known as Machado-Joseph Disease (MJD), is the most common form of autosomal dominant hereditary ataxia worldwide that mainly affects the cerebellum, brain stem and basal ganglia. It is clinically characterized by progressive ataxia and spasticity (Paulson et al., 2017). Like Huntington’s disease (HD), spinobulbar muscular atrophy (SBMA), and other spinocerebellar ataxias, including dentatorubral-pallidoluysian atrophy (DRPLA), SCA3/MJD is a polyglutamine (polyQ) expansion disease.
The genetic mutation is an expansion of a CAG trinucleotide repeat tract in the ATXN3 gene, which encodes ataxin-3. The CAG repeat length is 12–43 repeats in normal individuals and is expanded to 51–91 repeat units in SCA3/MJD patients (Saute and Jardim, 2015). In general, wild-type ataxin-3 is primarily a cytoplasmic protein; however, expanded ataxin-3 can translocate into the nucleus and form aggregates (Paulson et al., 1997; Schmidt et al., 1998). A growing amount of evidence has demonstrated that the nuclear environment is the primary site of pathology in SCA3 by promoting aggregate seeding (Fujigasaki et al., 2000; Sowa et al., 2018). Nuclear presence of ataxin-3 especially induces neurotoxicity, including the coaggregation of transcriptional factors, leading to transcriptional dysfunction and disturbing the nuclear organization and function and thereby inducing SCA3/MJD pathology (Sun et al., 2007; Bichelmeier et al., 2007; Breuer et al., 2010; Wang et al., 2018).
Disulfiram (chemical names: tetraethylthiuram disulfide or 1-(diethylthiocarbamoyldisulfanyl)-N,N-diethyl-methanethioamide; trade names: Antabuse or Antabus) is a Food and Drug Administration (FDA)-approved medication for chronic alcoholism. The mode of action for disulfiram is to block the enzyme aldehyde dehydrogenase (ALDH), which accumulates acetaldehyde and leads to a series of unfortunate reactions, which helps alcohol dependent patients stop drinking. It also shows a beneficial effect on various cancer types in preclinical investigations based on multiple pathways (Skrott et al., 2017). The most adverse effect of disulfiram treatment is neurotoxicity, including neurofilamentous distal axonopathy, sensorimotor polyneuropathy and peripheral neuropathy in human patients and experimental animal models (Kulkarni et al., 2013; Stone et al., 2014). In addition, disulfiram can cause mitochondrial membrane damage (Pavón et al., 2017). Interestingly, one of the pathogenic mechanisms of SCA3 is mitochondrial dysfunction (Duarte-Silva et al., 2018). Currently, the direct effect of disulfiram in SCA3 has not been investigated. Here, we investigate the FDA-approved drug disulfiram to assess whether it affects aggregations of ataxin-3, soluble mutant ataxin-3 and the nuclear localization of ataxin-3 and influences the cell viability in vitro.
Human Embryonic Kidney 293 (HEK293) cells were cultured as previously described (Antony et al., 2009). Chinese hamster ovary (CHO) cells were grown in F12 medium (Gibco, Life Technologies, Darmstadt, Germany) supplemented with 10% foetal bovine serum (Gibco, Life Technologies), 1% penicillin/streptomycin (Gibco, Life Technologies) and 300 µg/mL hygromycin B (Invitrogen, Carlsbad, CA, USA) in a humidified incubator at 37°C with 5% CO2.
Cell transfectionThe pEGFP-N2-Ataxin-3 constructs, including three different CAG repeats stretches (15, 77 or 148 CAG repeats), were used for cell transfection. CHO cells were stably transfected with pEGFP-N2-Ataxin-3-15CAG/77CAG plasmids using FuGENE 6 (Roche, Basel, Switzerland) according to the manufacturer’s protocol. The pEGFP-N2-Ataxin-3-148CAG/15CAG was used for transient transfection into HEK293 cells using the Attractene Transfection Reagent (QIAGEN, Hilden, Germany) according to the manufacturer’s procedure.
Protein extractionSubcellular fractionation was performed to isolate the cytoplasmic and nuclear protein fractions as previously described (Wang et al., 2018). For total protein extraction, cells were collected and extracted as before (Wang et al., 2018).
SDS PAGE and Western blotThe protein concentration was measured using a Bradford assay (Bradford, 1976; Bio-Rad Laboratories, Munich, Germany). SDS PAGE and Western blot were performed as previously described (Schmidt et al., 1998). Protein lysates were transferred onto nitrocellulose (NC) membranes (0.2 μm, GE Healthcare Life Sciences, Chicago, IL, USA). The membranes were subsequently blocked in 1 × TBST (10 mM Tris pH 7.5; 0.15 M NaCl; 0.1% Tween 20) with 4% (w/v) low-fat dry milk (SlimFast, Allpharm Vertriebs GmbH, Messel, Germany) for 2 hr and probed with the primary antibody diluted in 1 × TBST for 2 hr at room temperature or overnight at 4°C. After washing three times with 1 × TBST, the blot was incubated with the secondary antibody diluted in 1 × TBST for 1 hr, washed again, detected with an Amersham ECL Western Blotting Detection Kit (GE Healthcare Life Sciences) and processed using an Odyssey Fc system (LI-COR Biotechnology, Lincoln, NE, USA).
ImmunofluorescenceImmunofluorescence staining was carried out as described previously (Bichelmeier et al., 2007). The cells were plated at 2 × 105 cells/well in 6-well plates containing a poly-L-lysine coated cover slide and grown at 37°C overnight. The cells were treated with disulfiram for 2 hr on the following day. After removing the old medium, cells were fixed with an ice-cold mixture of methanol:acetone (1:1) for 5 min. The cells were subsequently washed with PBS, blocked with blocking buffer (DPBS + 3% normal donkey serum) for 20 min at room temperature. Then, the cells were incubated with a primary antibody for 1 hr or overnight at 4°C, washed with PBS three times and incubated with a secondary antibody diluted in blocking buffer for 1 hr at room temperature. After washing three times with PBS for 5 min each, cells were stained with DAPI (1:10,000 in PBS) for 10 min, washed again, then the cover slide was placed on a slide with 100 µL of mounting medium (freshly mixed mowiol (Merck, Kenilworth, NJ, USA) + 2.5% DABCO [1,4-diazabicyclo(2.2.2)octane]; (Sigma-Aldrich, Saint Louis, MO, USA)). Fluorescence intensity was detected with an Axioplan 2 imaging light microscope (Carl Zeiss, Oberkochen, Germany).
Filter trap assayFilter trap assays were carried out to detect the SDS-resistant insoluble aggregations of mutant ataxin-3 (Wanker et al., 1999). Seventy-two hours after transfection, HEK293 cells were collected, centrifuged at 250 × g for 10 min and resuspended in 300 µL of PBS with protease inhibitor (Roche). The lysates were sonicated for 1 min for cell lysis and DNA fragmentation. The protein concentration was measured as described before. One hundred micrograms of sample was supplemented with 2% SDS and loaded onto a cellulose acetate membrane (0.2 µm, GE Healthcare Life Sciences) on top of filter paper. The filter trap apparatus was run and washed with PBS. The membrane was then prepared for Western blot as described above.
Cell viability assayThe PrestoBlue Cell Viability Reagent (Life Technologies, Darmstadt, Germany) is a resazurin-based compound used for assessing cell viability. Cells were seeded at 5 × 104 cells/well in a 96-well plate and treated with disulfiram for 2 hr or 24 hr. Cell viability was then analysed with the PrestoBlue Cell Viability Reagent after incubation for 10 min and measurement by the fluorescent intensity using a Synergy HT microplate reader (excitation: 535 nm (25 nm bandwidth), emission: 615 nm (10 nm bandwidth; BioTek, Bad Friedrichshall, Germany).
Statistical analysesQuantification of data from three independent experiments is presented as the means ± standard error of the mean (SEM). Statistical analysis was performed using Student’s t-test for data with a normal distribution. The effect of multiple factors was tested by a two-way analysis of variance (ANOVA) test. Significance levels are described as follows: P < 0.05*, P < 0.01**, P < 0.001***, and P < 0.0001**** except where noted.
Considering the maximum daily dose of 500 mg of disulfiram for alcohol abuse, which is equivalent to 5 μM of disulfiram in the blood (Faiman et al., 1984), and the effective concentration of disulfiram in inhibiting proteasome (0.16-20 μM) (Chen et al., 2006; Lövborg et al., 2006), we intentionally performed the experiment using low dose of 10 µM to evaluate the safety of disulfiram in clinical application. The massive toxicity observed at higher concentrations should be avoided, also as incubation for 24 hr in experiments was required. Firstly, we investigated the effect of disulfiram on the aggregate formation of expanded ataxin-3, which plays a toxic role in SCA3. A filter trap assay was performed. We used HEK293 cells transfected with ataxin-3148CAG or ataxin-315CAG as a negative control and treated them with disulfiram and DMSO as a control. The filter trap assay revealed no significant difference between the disulfiram-treated cells and the DMSO-treated cells (Fig. 1), which suggests that disulfiram did not affect the aggregation at one single dose.

Filter traps assays showed that disulfiram did not alter the aggregate formation of mutant ataxin-3. HEK293 cells expressing ataxin-3148Q or ataxin-315Q were treated with disulfiram (Selleck Chemical, Munich, Germany) at 10 μM in DMSO or DMSO alone as a control for 24 hr. The signal was normalized to that of untreated cells. Student’s t-test.
The soluble mutant ataxin-3 also showed toxicity to the SCA3 cell model. To evaluate whether the toxicity of disulfiram is due to alteration in the level of soluble mutant ataxin-3, the protein levels of ataxin-3 were assessed using Western blot. Analysis of Western blot showed that disulfiram did not affect the protein level of ataxin-377Q or ataxin-315Q as a control (Fig. 2) in comparison with that in the DMSO-treated group. This result demonstrates that disulfiram at low dose did not alter the level of soluble mutant ataxin-3.

Disulfiram did not change the soluble protein levels of ataxin-315Q or ataxin-377Q. Soluble protein expression in CHO cells stably expressing ataxin-315Q or ataxin-377Q was analysed by Western blots with anti-ataxin-3 (1H9, Merck Millipore, Darmstadt, Germany) and anti-actin (loading control, Sigma Aldrich, St. Louis, MO, USA) antibodies (A and B, respectively). Cells were treated with disulfiram (at 10 μM in DMSO for 2 hr) or DMSO alone as a control. Western blots were analysed based on ataxin-315Q or ataxin-377Q signals normalized to actin signals as a loading control (C and D, respectively). Student’s t-test.
Ataxin-3 commonly shows more toxicity in the nucleus than in the cytoplasm (Bichelmeier et al., 2007; Sowa et al., 2018). Moreover, ataxin-3 nuclear accumulation is the earliest pathological change in SCA3. With the aim of investigating the role of disulfiram in the translocation of ataxin-3 from cytoplasm to nucleus in vitro, fractionation analysis and immunofluorescence assays were performed. Fractionation results showed increased nuclear localization of ataxin-377Q in disulfiram-treated cells but not of ataxin-315Q (Fig. 3). In addition, immunofluorescence assays confirmed a significantly increased nuclear localization of ataxin-377Q in disulfiram-treated cells in comparison with the localization in DMSO-treated cells (Fig. 4). In contrast, the distribution of ataxin-315Q was not changed.
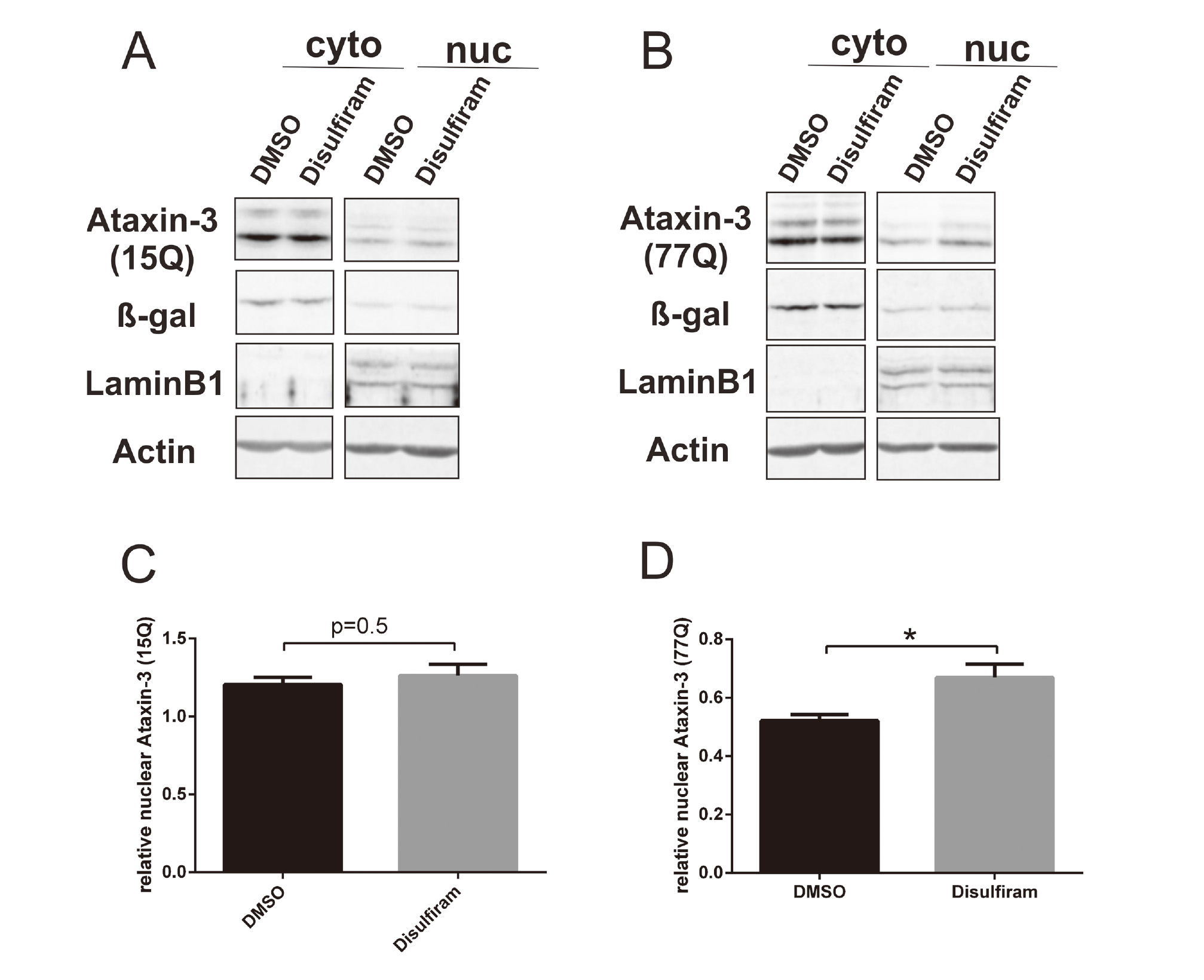
Disulfiram showed an increased nuclear localization of ataxin-377Q by subcellular fractionation. Cytoplasmic and nuclear fractions of CHO cells stably expressing ataxin-315CAG or ataxin-377CAG were analysed by Western blotting with anti-ataxin-3, anti-β-galactosidase (cytoplasmic marker, Rockland, Limerick, PA, USA), anti-Lamin B1 (nuclear marker, Santa Cruz Biotechnology, Santa Cruz, CA, USA), and anti-actin antibodies (A and B, respectively). Cells were treated with disulfiram (at 10 μM in DMSO for 2 hr) or DMSO alone as a control. Western blots were analysed based on ataxin-315Q or ataxin-377Q signals normalized to actin signals as a loading control. Western blots were densitometrically quantified as a percentage of nuclear ataxin-315Q or ataxin-377Q based on the total nuclear and cytoplasmic quantification (C and D, respectively). Student’s t-test. *p < 0.05.

Disulfiram significantly increased the nuclear localization of ataxin-377Q as shown by immunofluorescence assays. Treatment with disulfiram (at 10 μM in DMSO for 2 hr) significantly increased the nuclear localization of ataxin-377Q (A and C), while no significant changes in the localization of normal ataxin-3 were observed (B). The same CHO cells treated with disulfiram were stained with an anti-ataxin-3 antibody (green) and DAPI (blue, nuclear marker). At least 60 cells in each group were analysed (n ≥ 60). Scale bar: 20 μm. Student’s t-test. **p < 0.01.
Given that disulfiram has been reported to induce neurotoxicity, we tested the direct effect of disulfiram on the cytotoxicity of a cell model for SCA3. We used a resazurin-based Presto Blue reagent to assess whether disulfiram treatment reduced cell viability in ataxin-377CAG-transfected CHO cells. No cytotoxicity was observed in the presence of 10 μM disulfiram. However, decreased cell viability was observed when cells were exposed to a high concentration (100 µM) of disulfiram both at 2 and 24 hr (Fig. 5). This is the direct evidence that disulfiram could aggravate the cytotoxicity in a cell model of SCA3.

Disulfiram at a concentration of 100 µM showed cytotoxicity to the cells. CHO cells expressing ataxin-377Q were seeded in 96-well plates and treated with disulfiram or DMSO as a control at different concentrations for 2 hr (A) or 24 hr (B). The x-axis shows the log10 of the disulfiram concentration. The y-axis represents fluorescence values normalized to those of untreated cells. Two-way ANOVA. ****p < 0.0001.
Disulfiram is an FDA-approved drug used to treat chronic alcoholism by blocking acetaldehyde dehydrogenase. In addition, disulfiram is being used for cancer treatment in preclinical studies and clinical trials. Our previous study demonstrated that expanded ataxin-3 localization in the nucleus is more toxic than localization in the cytoplasm (Bichelmeier et al., 2007; Sowa et al., 2018). So far, there are very few compounds available that effectively influence the translocation of ataxin-3 into the nucleus (Mueller et al., 2009; Pastori et al., 2010; Wang et al., 2018). Disulfiram is an attractive “old” drug; however, our study identified its function as an inducer of ataxin-3’s nuclear translocation without affecting ataxin-3’s aggregation and showed its cytotoxicity.
How does it achieve this effect? Beyond treating alcoholism by blocking ALDH, disulfiram achieves its anticancer effects primarily by inhibition of the proteasome, by blocking NF-kB, and also by the generation of reactive oxygen species (ROS) (Cvek and Dvorak, 2008; Lövborg et al., 2006; Xu et al., 2017). The proteasome is responsible for the degradation of ubiquitinated mutant polyQ proteins (Schulman and Harper, 2009; Wade et al., 2014). Inhibitors of the proteasome cause the activation of caspases (pro-apoptotic proteins) and calpains and an increase in reactive oxygen species and ubiquitinated proteins in HD fibroblasts and other cell models (Li et al., 2008; Fernandez-Estevez et al., 2014). Caspases and calpains are both responsible for ataxin-3 proteolysis and contribute to the pathogenesis of SCA3 (Berke et al., 2004; Weber et al., 2017). Moreover, the proteolytic fragments produced by cleavage of mutant ataxin-3 lead to nuclear translocation of ataxin-3 and cell death. In addition, it has been reported that disulfiram induces oxidative stress (Grosicka-Maciąg et al., 2010; Pavón et al., 2017). Oxidative stress has also been characterized as an inducer of nuclear ataxin-3 transport (Reina et al., 2010). This may partially explain why disulfiram, a proteasome inhibitor, increases nuclear ataxin-3 in vitro. Recent evidence shows that disulfiram also plays a role in inhibiting human transglutaminase 2 (TG2) (Palanski and Khosla, 2018). In addition, transglutaminases (TGs) are multifunctional proteins that are involved in protein cross-linking and facilitating autophagic completion (Grosso and Mouradian, 2012). Importantly, inhibiting TG2 can increase the expanded ataxin-3-induced neurotoxicity in an SCA3 fly model (Lin et al., 2015). This may explain why disulfiram showed toxicity to cells expressing ataxin-3 and TG2 is potential pathway to explore for further studies. In addition, our data imply that the potential toxic effect of disulfiram may result from the influence of increasing the nuclear transport of ataxin-3.
Disulfiram is an “old” drug for alcoholism and a promising agent for cancer patients. However, current evidence indicates that disulfiram should only be considered with care in patients with SCA3. This study is also important to elucidating the intracellular mechanism controlling the localization of ataxin-3. Treatment with disulfiram had no significant effect on soluble ataxin-3 or on the aggregation of expanded ataxin-3. This could indicate that the concentration is efficient for influencing ataxin-3 translocation but not sufficient for aggregate formation as the next stage. Moreover, only 8-15% ataxin-3 (either subcellular fractionation assay or immunofluorescent method) in the cells treated with disulfiram was increased in the nucleus at a single dose. The larger number of samples, different methods, various doses and treatment times need to be further explored. Together, our study demonstrated that disulfiram should only be used with great care for the treatment of alcohol abuse or cancer in patients carrying an SCA3-causing mutation and that disulfiram triggers the nuclear transport of ataxin-3.
This work was supported by the Natural Science Basic Research Plan in Shaanxi Province of China (grant number 2017JQ8029); the Scientific Technology Program & Innovation Fund of Xi’an City Special Project (grant number 2016CXWL04); and Scientific research plan projects of Shaanxi Education Department (grant number 17JK1120). I would like to acknowledge Dr.ret.nat Thorsten Schmidt for providing the materials. I am also very grateful to Aoife hanet for her technical assistance and manuscript proofreading.
Conflict of interestThe authors declare that there is no conflict of interest.