Abstract
Long-term exposure to certain volatile organic compounds is a significant public health concern. A variety of food containers and drinking cups prepared from polystyrene or polystyrene-related plastics could contain styrene monomer. In the current study, the concentrations of styrene in plasma and liver were surveyed and determined after oral doses of 25 mg/kg to rats and 200 mg/kg to control and humanized-liver mice. Plasma concentrations of styrene in rats were still detected 2 hr after 10-25 mg/kg oral doses. In contrast, after an order of magnitude higher oral dose of styrene (200 mg/kg) to mice, styrene in mouse plasma was rapidly cleared within 15 min to the limit-of-detection level. However, unmetabolized styrene was detected in mouse liver 24 hr after oral treatment. A simple physiologically based pharmacokinetic (PBPK) model capable of estimating blood and liver concentrations of styrene was established for rats. A human PBPK model was then set up for styrene by using the same intrinsic hepatic clearances in rats and humans and by applying allometric scaling to rat parameters obtained from the plasma concentrations of styrene in rats. By reverse dosimetry analysis (from concentrations to doses), we found that the 95th percentile values of styrene concentrations (0.132 ng/mL) reported in United States biomonitoring data of more than 1000 human blood samples may imply exposure to repeated oral doses of styrene of 2.89 µg/kg/day. These results suggest that styrene biomonitoring data in human blood samples imply exposures roughly similar to or lower than the established tolerable daily intake level of 7.7 μg/kg/day.
INTRODUCTION
Volatile organic compounds originating from many different natural and man-made sources (Alwis et al., 2012) are ubiquitous in the human environment. Because long-term exposure to certain volatile organic compounds may increase the risk for cancer or birth defects, estimation of exposures of volatile organic compounds, including styrene, a group 2B human carcinogen (IARC, 2002), is an area of significant public health concern (Sexton et al., 2006). At present, epidemiologic evidence does not suggest a causal association between styrene and any forms of cancer in humans (Rueff et al., 2009). However, a variety of food containers, drinking cups, and carpets fabricated from polystyrene or polystyrene-related plastics could contain styrene monomer (Withey, 1976; Niaz et al., 2017). To predict the toxicokinetics of volatile chemicals in mixtures, integrated physiologically based pharmacokinetic (PBPK) modelling has been proposed (Krishnan and Johanson, 2005; Ramsey and Andersen, 1984; Kirman et al., 2003). Moreover, because the lung is considered the relevant target organ for styrene-induced carcinogenicity in mice, a full physiological toxicokinetic model has been developed to simulate the lung burden of styrene (Csanády et al., 2003).
In the mouse, levels of unmetabolized styrene in subcutaneous adipose tissue and liver seemed to increase exponentially with the dose (Löf et al., 1983). Styrene administration to mice has been shown to rapidly decrease glutathione levels in the liver and lung (Turner et al., 2005). Styrene is metabolically activated via its primary metabolite, styrene 7,8-epoxide (Löf et al., 1984); a role for cytochrome P450 (P450) 2E1 in styrene epoxidation has been reported (Hartman et al., 2015; Hartman et al., 2013; Hartman et al., 2012). Interestingly, DNA adduct formation reportedly does not play an important role in styrene tumorigenicity in chronically exposed mice (Boogaard et al., 2000). With regard to humans, P450 2A13, mainly located in the respiratory tract, efficiently catalyzed styrene 7,8-oxide formation from styrene (Fukami et al., 2008). The main urinary excretion products of styrene have been reportedly used in monitoring workspace exposure (Takeuchi et al., 2019). Low levels of styrene in human blood samples have been reported in the Fourth National Report on Human Exposure to Environmental Chemicals by the US Centers for Disease Control and Prevention. To date, forward and reverse dosimetry assessments of blood styrene levels have not been carried out in humans.
The aim of the present this study was to conduct forward and reverse dosimetry assessments of styrene levels by establishing a human PBPK model based on rodent data (Withey, 1976). We report herein that styrene biomonitoring data based on human blood samples imply exposures roughly similar to or lower than the tolerable daily intake (TDI) level of 7.7 μg/kg/day.
MATERIALS AND METHODS
In vivo and in vitro experiments
Male Sprague-Dawley rats (7 weeks old, Charles River, Yokohama, Japan), male ICR mice (7 weeks old, Charles River), and male TK-NOG mice and humanized-liver TK-NOG mice (25 g, Central Institute for Experimental Animals, Kawasaki, Japan; Hasegawa et al., 2011) were orally treated with styrene (25 or 200 mg/kg, Fujifilm Wako Pure Chemicals, Osaka, Japan) to determine the plasma and/or hepatic concentrations. The use of animals in this study was approved by the Ethics Committees of the Central Institute for Experimental Animals, Shin Nippon Biomedical Laboratories, and Showa Pharmaceutical University. Liver microsomes from 7-week-old male rats were prepared as described previously (Yamashita et al., 2014). The in vitro elimination rates of styrene mediated by liver microsomes from three individual rats and by pooled human liver microsomes (H150, Corning, Woburn, MA, USA) were calculated. Typical incubation mixtures contained an NADPH-generating system, potassium phosphate buffer (100 mM, pH 7.4), liver microsomes (0.25 mg protein/mL), and styrene (10 µM) in a final volume of 1.0 mL. To evaluate the elimination of styrene within the linear range, incubations were carried out for 30 min at 37°C. In vitro hepatic intrinsic clearance (CLh,int,in vitro) values in rats were estimated from the styrene elimination rates and were extrapolated to humans by applying the following values: 1 g liver contains 40 mg liver microsomal protein, and 40 g of liver weight per kilogram of rat bodyweight and 21.4 g of liver weight per kilogram of human bodyweight (Takano et al., 2010).
Liquid chromatography-tandem mass spectrometry assays
The concentrations of styrene in plasma and/or liver after oral administration with styrene were determined using liquid chromatography (LC)-tandem mass spectrometry. Plasma samples (20 µL) were deproteinized by adding 80 µL methanol (1:1, v/v) including styrene-d5 (7.5 µg/mL) followed by centrifugation (4°C, 1600 × g, 5 min) to obtain the supernatant as an LC sample. Mouse liver homogenates were prepared with an equal amount of 0.5 M potassium phosphate buffer (pH 6.0) and were extracted with two volumes of acetonitrile followed by centrifugation (4°C, 2500 × g, 15 min). Aliquots of the in vivo and in vitro LC samples were injected into a liquid chromatography 20A System (Shimadzu, Kyoto, Japan) equipped with an octadecylsilane (C18) column (YMC-Triart C18 plus: 3 µm, 2.1 mm × 50 mm, YMC, Kyoto, Japan) under the following conditions: a column temperature of 40°C; a mobile phase A of 0.1% acetic acid aqueous solution and solvent B of methanol; a gradient program of 0-2 min 70-90% of B, 2.1-4 min 100% of B, and 4.1-6 min 70% of B; and a flow rate of 0.3 mL/min. The mass spectra were obtained with a Triple Quad 6500 system (AB SCIEX, Framingham, MA, USA) under the following conditions: atmospheric pressure chemical ionization; multiple reaction monitoring; positive ion detection; and a capillary temperature of 600°C. Styrene and styrene-d5 were quantified using the m/z 104.2→103.1 transition and the m/z 109.2→108.1 transition, respectively.
Estimation of plasma and liver concentrations using simplified pharmacokinetic models
Simplified PBPK models for styrene (molecular weight: 104.15) consisted of a chemical receptor (gut) compartment, a metabolizing (liver) compartment, and a central compartment with a peripheral compartment and were established as previously described (Kamiya et al., 2019; Yamazaki et al., 2016). The octanol-water partition coefficient (logP: 2.86) and the plasma unbound fraction (fu,p: 0.136) of styrene were estimated in silico using ChemBioDraw and Simcyp (Emoto et al., 2009). The blood-to-plasma concentration ratio (Rb: 1.0) and the liver-to-plasma concentration ratio (Kp,h: 2.57) were calculated from fu,p and logP values (Poulin and Theil, 2002). Physiological values such as the hepatic blood flow rate (Qh) for rats (0.853 L/h) and humans (96.6 L/h) were obtained from the literature (Kato et al., 2008).
The concentrations of styrene in plasma were also taken from the literature (Withey, 1976). The volume of the systemic circulation (V1), the absorption rate constant (ka), and the in vivo hepatic intrinsic clearance (CLh,int) were evaluated using nonlinear regression analysis fitting techniques, as described previously (Kamiya et al., 2019). The renal clearance (CLr) was set at a minimum value compared with the hepatic clearance (CLh). Table 1 shows the final parameter values that apply to the rat PBPK model. Finally, to conduct the modeling for styrene, we solved the following system of differential equations (Takano et al., 2010):

where Xg and Xperipheral are the amounts of the chemicals in the gut and peripheral compartments respectively; Ch and Cb are the hepatic and blood chemical concentrations, respectively; Vh and V1 are the volumes of the liver and the central compartment, respectively; and Qh is the hepatic blood flow rate of systemic circulation to the hepatic compartment. The parameters required for the simplified human PBPK model for styrene were determined from the parameters of the rat PBPK model, from physiological parameters derived from the literature, and from the results of the liver microsomal experiments. The values of ka, V1, and CLh, for styrene in humans were estimated using a scale-up strategy from rats to humans, as described previously (Takano et al., 2010; Yamazaki et al., 2016; Kamiya et al., 2019). Briefly, the human ka value was obtained by multiplying the rat absorption rate constant (ka) by 0.744; the human systemic circulation volume (V1,human) was estimated using Vh and the blood volume (Vb), with Vh,human, Vb,rat, and Vb,human values of 1.50, 0.0160, and 4.90 L, respectively (Yamashita et al., 2014):
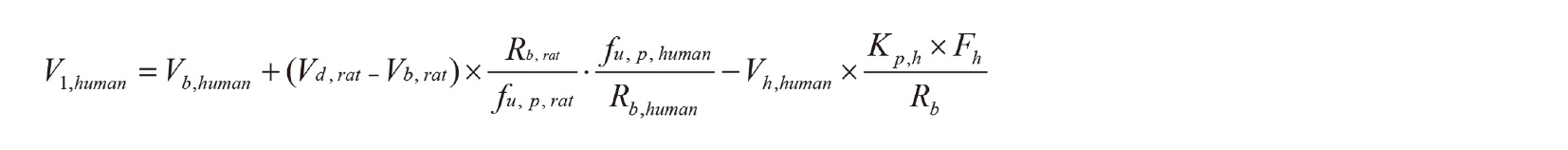
where Vd is the volume of distribution per unit body weight and Fh is the unmetabolized fraction in liver. The human in vivo hepatic intrinsic clearance (CLh,int) was estimated according to the methods of Takano et al. (2010) and Yamashita et al. (2014) by multiplying the initial calculated parameters for human in vitro hepatic intrinsic clearance (1.5 L/h) by the in vivo to in vitro hepatic intrinsic clearance ratio for styrene (~1) in rats. Table 1 gives the final parameter values for the human PBPK model. The system of differential equations described above was then solved to estimate the concentrations of styrene in each compartment in humans. The modeled plasma concentration curves of styrene for humans were compared with the 95th percentile concentrations for men reported by the US Centers for Disease Control and Prevention. Styrene levels in liver fractions from control and humanized-liver mice were statistically compared using an unpaired t-test with Prism (GraphPad Software, San Diego, CA, USA).
Table 1. Physiological and experimental parameters for rat and human PBPK models for styrene.
| Parameter |
Abbrev. (unit) |
Rat |
Human (from rat) by a scale-up strategy |
| Absorption rate constant |
ka (1/h) |
24.0 (± 0.5) |
17.8 |
| Fraction absorbed × intestinal availability |
Fa·Fg |
0.91 |
1 |
| Transfer rate constant |
k12 (1/h) |
2.56 (± 0.30) |
1.91 |
| Transfer rate constant |
k21 (1/h) |
1.69 (± 0.31) |
1.26 |
| Volume of systemic circulation |
V1 (L) |
0.0620 (± 0.0010) |
6.90 |
| Hepatic intrinsic clearance |
CLh,int (L/h) |
14.5 (± 0.3) |
466 |
| Hepatic clearance |
CLh (L/h) |
0.595 |
38.3 |
| Renal clearance |
CLr (L/h) |
0.001 |
0.0428 |
Data in the first three rows for the rat are means ± standard deviations. Parameters such as Kp,h, Rb, and fu,p were assumed to be the same for humans and for rats.
RESULTS AND DISCUSSION
In this study, the plasma concentrations of styrene were measured after oral doses of 25 mg/kg to rats and 200 mg/kg to control and humanized-liver mice. In rats, the dose-normalized plasma concentrations of styrene after 25 mg/kg oral doses in the present study were very similar in the elimination phase (Fig. 1A) to reported plasma concentrations of styrene after administration of 10 mg/kg (Withey, 1976). However, when an order of magnitude higher oral dose of styrene (200 mg/kg) was administered to mice, styrene in plasma from four individual ICR mice, from control TK-NOG mice, and from humanized-liver mice was rapidly cleared within 15 min to levels at the limit of detection (0.1 µg/mL, Table 2). In contrast, unmetabolized styrene was detected in the liver of these mice 24 hr after oral treatment (Fig. 1B). The mean hepatic concentrations of styrene in control and humanized-liver mice were 16.3 and 53.0 ng/g liver, respectively; these values were apparently different, but the difference was not significant, with a p value of 0.052 by the unpaired t-test.

Table 2. Plasma concentrations of styrene in four individual mice after oral doses of 200 mg/kg to ICR, TK-NOG, and humanized-liver mice.
| Time after administration, min |
ICR mice |
TK-NOG mice |
Humanized-liver mice |
| µg/mL |
| 5 |
0.71, 0.52, < 0.1, < 0.1 |
0.25, < 0.1, < 0.1, < 0.1 |
0.68, 0.30, < 0.1, < 0.1 |
| 15 |
< 0.1, < 0.1, < 0.1, < 0.1 |
0.32, < 0.1, < 0.1, < 0.1 |
0.10, < 0.1, < 0.1, < 0.1 |
Based on the reported plasma concentrations of styrene in rats after administrations of 10 mg/kg (Withey, 1976), a rat PBPK model was set up using the parameters shown in Table 1. The modeled results after virtual oral administration showed a good fit with the reported concentrations (the line in Fig. 1A). In vitro hepatic intrinsic clearances (CLh,int, in vitro) in rats and humans were determined from the liver microsome elimination rates (71 and 81 µL/min/mg protein, respectively, Table 3). An allometric scaling approach was applied to rat parameters without considering any interspecies factors for intrinsic hepatic clearances in rats and humans. Using these methods, the parameters for the human PBPK model were established for styrene (Table 1). By solving the equations of the simplified human PBPK model, in silico plasma and liver concentration curves after virtual administration of styrene were generated and are shown in Fig. 1C.
Table 3. Styrene elimination rates by liver microsomes from three individual rats and pooled human liver microsomes.
| Enzyme source |
Metabolic disappearance of styrene, pmol/min/mg protein |
Clearance, µL/min/mg protein |
| Rat liver microsomes |
|
|
| No. 1 |
694 |
69 |
| No. 2 |
714 |
71 |
| No. 3 |
724 |
72 |
| Mean and SD |
711 ± 16 |
71 ± 2 |
| Pooled human liver microsomes |
814 |
81 |
Styrene (10 µM) was incubated with liver microsomes (0.25 mg/mL) for 30 min.
The present human PBPK model can estimate plasma and/or liver concentrations of styrene for virtual doses using forward dosimetry (i.e., estimating concentrations from doses), but it is also able to perform reverse dosimetry (i.e., estimating doses from concentrations). Using reverse dosimetry, we estimated the styrene doses required to generate certain plasma concentrations (Fig. 2). This analysis was based on the 95th percentile values of blood styrene concentrations [0.132 ng/mL (95% confidence interval 0.106-0.183)] from reported biomonitoring data of 1320 human blood samples taken in 2009-2010, as published by the US Centers for Disease Control and Prevention (Fig. 2). Based on the assumption that the reported blood styrene concentrations were steady state values, the 95th percentile values imply exposure to repeated oral doses of styrene of 2.89 µg/kg/day (95% confidence interval 2.32-4.01 µg/kg/day) for 14 days. Although the 50th percentile value of styrene concentrations has been reported as the limit of detection (< 0.03 ng/mL), this would imply exposure to repeated oral styrene doses of less than 0.658 µg/kg/day. A human TDI level of 7.7 μg/kg/day for styrene has been established by WHO (1996). Unmetabolized styrene in human blood samples is clearly present in some members of the general population. PBPK modeling in humans indicated high liver accumulations of styrene with low plasma concentrations, as shown in Fig. 1C. Similarly, Löf et al. (1983) reported high concentrations in subcutaneous adipose tissue and liver in the mouse. Therefore, the reported biomonitoring levels of styrene in human blood may imply much higher levels of styrene in the liver. Taken together, the finding a measurable amount of styrene in human blood samples might imply a role of the liver as styrene stock for blood in people who have been exposed to higher levels of styrene.

In conclusion, in-depth forward and reverse dosimetry assessments of styrene levels were facilitated by adopting the human PBPK model established in this study. Using the model, we evaluated the biomonitoring data for styrene in human blood samples and found that the styrene levels imply exposures roughly similar to or lower than the TDI level of 7.7 μg/kg/day.
ACKNOWLEDGMENTS
This work was supported in part by the METI Artificial Intelligence-based Substance Hazard Integrated Prediction System Project, Japan. We thank David Smallbones for copyediting a draft of this article.
Conflict of interest
The authors declare that there is no conflict of interest.
REFERENCES
- Alwis, K.U., Blount, B.C., Britt, A.S., Patel, D. and Ashley, D.L. (2012): Simultaneous analysis of 28 urinary VOC metabolites using ultra high performance liquid chromatography coupled with electrospray ionization tandem mass spectrometry (UPLC-ESI/MSMS). Anal. Chim. Acta, 750, 152-160.
- Boogaard, P.J., de Kloe, K.P., Wong, B.A., Sumner, S.C., Watson, W.P. and van Sittert, N.J. (2000): Quantification of DNA adducts formed in liver, lungs, and isolated lung cells of rats and mice exposed to (14)C-styrene by nose-only inhalation. Toxicol. Sci., 57, 203-216.
- Csanády, G.A., Kessler, W., Hoffmann, H.D. and Filser, J.G. (2003): A toxicokinetic model for styrene and its metabolite styrene-7,8-oxide in mouse, rat and human with special emphasis on the lung. Toxicol. Lett., 138, 75-102.
- Emoto, C., Murayama, N., Rostami-Hodjegan, A. and Yamazaki, H. (2009): Utilization of estimated physicochemical properties as an integrated part of predicting hepatic clearance in the early drug-discovery stage: impact of plasma and microsomal binding. Xenobiotica, 39, 227-235.
- Fukami, T., Katoh, M., Yamazaki, H., Yokoi, T. and Nakajima, M. (2008): Human cytochrome P450 2A13 efficiently metabolizes chemicals in air pollutants: naphthalene, styrene, and toluene. Chem. Res. Toxicol., 21, 720-725.
- Hartman, J.H., Boysen, G. and Miller, G.P. (2012): CYP2E1 metabolism of styrene involves allostery. Drug Metab. Dispos., 40, 1976-1983.
- Hartman, J.H., Boysen, G. and Miller, G.P. (2013): Cooperative effects for CYP2E1 differ between styrene and its metabolites. Xenobiotica, 43, 755-764.
- Hartman, J.H., Letzig, L.G., Roberts, D.W., James, L.P., Fifer, E.K. and Miller, G.P. (2015): Cooperativity in CYP2E1 metabolism of acetaminophen and styrene mixtures. Biochem. Pharmacol., 97, 341-349.
- Hasegawa, M., Kawai, K., Mitsui, T., Taniguchi, K., Monnai, M., Wakui, M., Ito, M., Suematsu, M., Peltz, G., Nakamura, M. and Suemizu, H. (2011): The reconstituted ‘humanized liver’ in TK-NOG mice is mature and functional. Biochem. Biophys. Res. Commun., 405, 405-410.
- International Agency For Research On Cancer (IARC) (2002): Overall Evaluations Of Carcinogenicity: An Updating Of IARC Monographs Volume 82, Lyon, P 437.
- Kamiya, Y., Otsuka, S., Miura, T., Takaku, H., Yamada, R., Nakazato, M., Nakamura, H., Mizuno, S., Shono, F., Funatsu, K. and Yamazaki, H. (2019): Plasma and Hepatic Concentrations of Chemicals after Virtual Oral Administrations Extrapolated Using Rat Plasma Data and Simple Physiologically Based Pharmacokinetic Models. Chem. Res. Toxicol., 32, 211-218.
- Kato, M., Shitara, Y., Sato, H., Yoshisue, K., Hirano, M., Ikeda, T. and Sugiyama, Y. (2008): The quantitative prediction of CYP-mediated drug interaction by physiologically based pharmacokinetic modeling. Pharm. Res., 25, 1891-1901.
- Kirman, C.R., Sweeney, L.M., Meek, M.E. and Gargas, M.L. (2003): Assessing the dose-dependency of allometric scaling performance using physiologically based pharmacokinetic modeling. Regul. Toxicol. Pharmacol., 38, 345-367.
- Krishnan, K. and Johanson, G. (2005): Physiologically-based pharmacokinetic and toxicokinetic models in cancer risk assessment. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev., 23, 31-53.
- Löf, A., Gullstrand, E. and Byfält Nordqvist, M. (1983): Tissue distribution of styrene, styrene glycol and more polar styrene metabolites in the mouse. Scand. J. Work Environ. Health, 9, 419-430.
- Löf, A., Gullstrand, E., Lundgren, E. and Nordqvist, M.B. (1984): Occurrence of styrene-7,8-oxide and styrene glycol in mouse after the administration of styrene. Scand. J. Work Environ. Health, 10, 179-187.
- Niaz, K., Mabqool, F., Khan, F., Ismail Hassan, F., Baeeri, M., Navaei-Nigjeh, M., Hassani, S., Gholami, M. and Abdollahi, M. (2017): Molecular mechanisms of action of styrene toxicity in blood plasma and liver. Environ. Toxicol., 32, 2256-2266.
- Poulin, P. and Theil, F.P. (2002): Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism-based prediction of volume of distribution. J. Pharm. Sci., 91, 129-156.
- Ramsey, J.C. and Andersen, M.E. (1984): A physiologically based description of the inhalation pharmacokinetics of styrene in rats and humans. Toxicol. Appl. Pharmacol., 73, 159-175.
- Rueff, J., Teixeira, J.P., Santos, L.S. and Gaspar, J.F. (2009): Genetic effects and biotoxicity monitoring of occupational styrene exposure. Clin. Chim. Acta, 399, 8-23.
- Sexton, K., Adgate, J.L., Fredrickson, A.L., Ryan, A.D., Needham, L.L. and Ashley, D.L. (2006): Using biologic markers in blood to assess exposure to multiple environmental chemicals for inner-city children 3-6 years of age. Environ. Health Perspect., 114, 453-459.
- Takano, R., Murayama, N., Horiuchi, K., Kitajima, M., Kumamoto, M., Shono, F. and Yamazaki, H. (2010): Blood concentrations of acrylonitrile in humans after oral administration extrapolated from in vivo rat pharmacokinetics, in vitro human metabolism, and physiologically based pharmacokinetic modeling. Regul. Toxicol. Pharmacol., 58, 252-258.
- Takeuchi, A., Namera, A., Sakui, N., Yamamoto, S., Yamamuro, K., Nishinoiri, O., Endo, Y. and Endo, G. (2019): Direct methyl esterification with 2,2-dimethoxypropane for the simultaneous determination of urinary metabolites of toluene, xylene, styrene, and ethylbenzene by gas chromatography-mass spectrometry. J. Occup. Health, 61, 82-90.
- Turner, M., Mantick, N.A. and Carlson, G.P. (2005): Comparison of the depletion of glutathione in mouse liver and lung following administration of styrene and its metabolites styrene oxide and 4-vinylphenol. Toxicology, 206, 383-388.
- WHO. (1996): Guidelines For Drinking-Water Quality, 2nd Ed. Vol. 2. Health Criteria And Other Supporting Information. Geneva, World Health Organization, 1996. Pp. 486-494.
- Withey, J.R. (1976): Quantitative analysis of styrene monomer in polystyrene and foods including some preliminary studies of the uptake and pharmacodynamics of the monomer in rats. Environ. Health Perspect., 17, 125-133.
- Yamashita, M., Suemizu, H., Murayama, N., Nishiyama, S., Shimizu, M. and Yamazaki, H. (2014): Human plasma concentrations of herbicidal carbamate molinate extrapolated from the pharmacokinetics established in in vivo experiments with chimeric mice with humanized liver and physiologically based pharmacokinetic modeling. Regul. Toxicol. Pharmacol., 70, 214-221.
- Yamazaki, H., Suemizu, H., Mitsui, M., Shimizu, M. and Guengerich, F.P. (2016): Combining Chimeric Mice with Humanized Liver, Mass Spectrometry, and Physiologically-Based Pharmacokinetic Modeling in Toxicology. Chem. Res. Toxicol., 29, 1903-1911.