Original Article
Catecholamine oxidation-mediated transcriptional inhibition in Mn neurotoxicity
2020 Volume 45 Issue 10 Pages 619-624
Details
2020 Volume 45 Issue 10 Pages 619-624
Manganese (Mn) poisoning may result in a neurological disorder called manganism. Although the neurotoxic mechanism of Mn is unclear, oxidative stress may be involved based on the interactions between neurotransmitter catecholamines and metals such as iron. Here, we propose a novel mechanism in which Mn oxidizes catecholamines and inhibits cellular transcription. Mn accelerated the oxidation of adrenaline (Ad) and produced adrenochrome (AdC) more effectively than iron. Furthermore, the oxidation of DNA bases increased when Ad, Mn, and iron were present. However, despite the absence of iron, cell viability decreased in the presence of AdC or Ad with Mn, which suggests there is another mechanism independent of oxidative DNA damage. AdC or preincubated Ad with Mn reduced mRNA synthesis in T7 RNA polymerase-driven transcription. RNA synthesis decreased in AdC-treated cells dose-dependently. These results show that Mn disrupts neuronal function via catecholamine oxidation-mediated transcriptional inhibition.
Manganese (Mn) is an essential trace element for humans; however, excess intake leads to toxicity called ‘manganism’. In addition to occupational exposure, a toxicological relationship is also a matter of concern in those whose homeostasis is dysregulated (Ejima et al., 1992). Mn intoxication results in a neurological syndrome with partial overlapping clinical features of neurodegenerative diseases such as Parkinson’s disease (Lucchini et al., 2009). High Mn content in the scalp of patients with amyotrophic lateral sclerosis has been reported in the Kii Peninsula, Wakayama, Japan (Kihira et al., 2015). A common cause of neurodegenerative diseases is an increase in oxidative stress by certain metals such as iron (Fe) (Farina et al., 2013). Mn enhances the oxidation of catecholamines, catalyzes the production of hydrogen peroxide, and causes oxidative DNA damage in association with Fe or copper (Ando et al., 2011; Oikawa et al., 2006).
In addition to DNA damage leading to neuronal cell death, inhibition of DNA transcription may be important in neurotoxic consequences (Hetman et al., 2010). We previously showed that transcriptional inhibition resulted from oxidative DNA damage induced by dopamine plus copper or Fe (Nishino et al., 2011). Transcriptional inhibition-triggered p53 response may be caused not only by damage of template DNA but also by chemical inhibitors against RNA polymerases (Ljungman et al., 1999). Therefore, in addition to oxidative stress by transition metals, the resulting products of catecholamine may cause neural disorders via transcriptional inhibition. An oxidation product of catecholamine, aminochrome, may be relevant to neurodegenerative diseases with unknown functions (Segura-Aguilar, 2017). Here, we show Mn-induced transcriptional inhibition, which is mediated by an aminochrome rather than oxidative DNA damage. These results imply a novel mechanism for disorders of the catecholaminergic neuronal system.
Adrenaline (Ad), adrenochrome (AdC), 2′-deoxyguanosine (dG), 8-hydroxy-2′-deoxyguanosine (8-oxodG), and salmon sperm DNA were purchased from Sigma (St. Louis, MO, USA). Manganese(II) chloride [Mn(II)] was obtained from Nacalai Tesque (Kyoto, Japan). Iron(III) nitrate nonahydrate [Fe(III)], formaldehyde, ethylenediamine-N,N,N′,N′-tetraacetic acid disodium salt (EDTA), Dulbecco’s modified Eagle’s medium (DMEM), penicillin, and streptomycin were purchased from Wako Pure Chemical Industries (Osaka, Japan). Nitrilotriacetic acid trisodium salt (NTA) was obtained from Tokyo Kasei Kogyo (Tokyo, Japan). Fe(III) was used in an NTA-complex [Fe(III)-NTA] by mixing with an equimolar amount of NTA in aqueous solution. Nuclease P1 was purchased from Yamasa Shoyu (Chiba, Japan). Fetal bovine serum (FBS) from Nichirei Bioscience, Inc. (Tokyo, Japan) was used. Hind III and calf intestine alkaline phosphatase (CIP) were purchased from Roche Diagnostics (Mannheim, Germany). 5-Ethynyl uridine (EU) was purchased from Molecular Probes (Thermo Fisher Scientific, Waltham, MA, USA).
Measurement of Ad and AdCReaction mixtures containing Ad (10 μM) alone or Ad plus Mn(II) (20 μM), Fe(III)-NTA (20 μM), or both Mn(II) and Fe(III)-NTA in sodium phosphate buffer (4 mM, pH 7.8) were incubated at 37°C for 2 hr. The concentrations of Ad and AdC were determined by measuring absorbance at 279 and 488 nm, respectively, as previously reported (Remião et al., 2003). Conditions of high performance liquid chromatography (HPLC, LC-10 series, Shimadzu, Kyoto Japan) were as follows: column, cosmosil 5C18-MS-II (Nacalai Tesque, Inc.); column temperature, 37°C; mobile phase, 95% ammonium acetate buffer (ammonium acetate 1.4 g in 1900 mL water, pH 2.5) and 5% acetonitrile; flow rate, 0.75 mL/min.
Measurement of 8-oxodG formationSalmon sperm DNA (100 µM base) was treated with 10 µM AdC, 10 µM Ad, 1 µM Mn(II), and 20 µM Fe(III)-NTA in 400 µL of 4 mM sodium phosphate buffer (pH 7.8) at 37°C for 2 hr. The base oxidation was determined as previously described (Ueda et al., 2014). Briefly, DNA was dissolved in 20 mM sodium acetate buffer (pH 5.0) after ethanol precipitation and digested to nucleosides by incubating first with nuclease P1 (3.4 units) at 37°C for 30 min then CIP (1.3 units) at 37°C for 1 hr in 0.1 M Tris-HCl (pH 7.5). The mixtures were analyzed by HPLC equipped with both a UV detector (SPD-M10A VP, Shimadzu) and an electrochemical detector (ECD, Coulochem II, ESA, Chelmsford, MA, USA). The molar ratio of 8-oxodG to dG was measured based on the peak height of authentic 8-oxodG with ECD and UV absorbance at 254 nm of dG.
Measurement of cell viabilityA human neuroblastoma-derived cell line, SH-SY5Y, purchased from American Type Culture Collection (ATCC), was maintained in DMEM/Ham’s F-12 containing 10% FBS, 100 units/mL penicillin, and 100 μg/mL streptomycin at 37°C under humidified atmosphere with 5% CO2. For the viability assay, cells were inoculated at 1.0 × 104 cells/well on a 96-well culture plate and incubated for 24 hr. One-tenth volume of aqueous solution of 200 μM AdC, 200 μM Ad, 400 μM Mn(II), and 200 μM Fe(III)-NTA were added and incubated for 6 hr. Cell viability was evaluated using a resazurin-based reagent (PrestoBlue, Thermo Fisher Scientific) according to the manufacturer’s instructions. Fluorescence was measured at an excitation wavelength of 544 nm and emission wavelength of 612 nm using a plate reader (Gemini EM, Molecular Devices, Sunnyvale, CA, USA) and shown as percent of untreated cells.
In vitro transcription assayIn vitro transcription was performed as described previously (Ueda et al., 2014) using a reconstituted transcription system (ScriptMAX Thermo T7 transcription kit, Toyobo, Osaka, Japan). Template DNA (pTD-Gal, Ezure et al., 2010) was prepared from Escherichia coli cell culture and linearized with Hind III. The reaction system (20 µL) contained 2 µL of 25-200 µM Ad or AdC, or 2 mM Ad and 1-1000 µM Mn(II) preincubated for 2 hr at 37°C. The reaction mixture was then mixed with RNA loading buffer AG+ (Biodynamics Laboratory Inc., Tokyo, Japan) and formaldehyde, denatured according to the instruction manual, and subjected to nondenaturing 1% agarose gel electrophoresis. Migrated RNA was quantified by a fluorescent imager (Typhoon FLA 9000, GE Healthcare, Uppsala, Sweden).
Measurement of cellular RNA synthesisHuman breast cancer cell line, MCF-7, purchased from the European Collection of Cell Cultures (ECACC, Salisbury, UK), was maintained in DMEM (low glucose) supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were inoculated at 3.0 × 105 cells/well on a 35-mm dish and incubated for 24 hr. EU (1 mM) and different concentrations of AdC were added and incubated for 24 hr. Total RNA was prepared from the cells using a kit (RNeasy, Qiagen GmbH, Hilden, Germany), and its concentration was measured using a UV-vis spectrophotometer (NanoPhotometer, Implen GmbH, München Germany). Newly synthesized RNA was detected by labeling incorporated EU with azide-modified fluorescent dye according to the instruction manual (Click-iT RNA Alexa Fluor 488 Imaging Kit, Thermo Fisher Scientific). The labeled RNA was purified by repeating ethanol precipitation twice to remove unreacted fluorescent dye. Fluorescence was measured with excitation at 495 nm and emission at 519 nm using a microplate reader (FlexStation 3, Molecular Devices, San Jose, CA, USA).
StatisticsData are shown as the mean ± S.D. of three separate experiments for Ad oxidation, 8-oxodG formation, cytotoxicity, and inhibition of T7-transcription. Difference compared with the control was analyzed by t-test and a noticeable tendency was described in the text.
We clarified the effect of Mn on the oxidation of Ad to AdC under conditions near physiological pH. Ad and AdC were monitored every 30 min by measuring maximum absorption at 279 and 488 nm, respectively. Ad was stable in the absence of metals and remained at more than 60% of the initial amount in the presence of Fe for up to 120 min. In contrast, Ad was almost depleted in 120 min in the presence of Mn and in less than 90 min when both Fe and Mn were present (Fig. 1A). In the same mixture, the concentration of AdC increased, which was consistent with the rate of decrease in Ad in each condition (Fig. 1B).
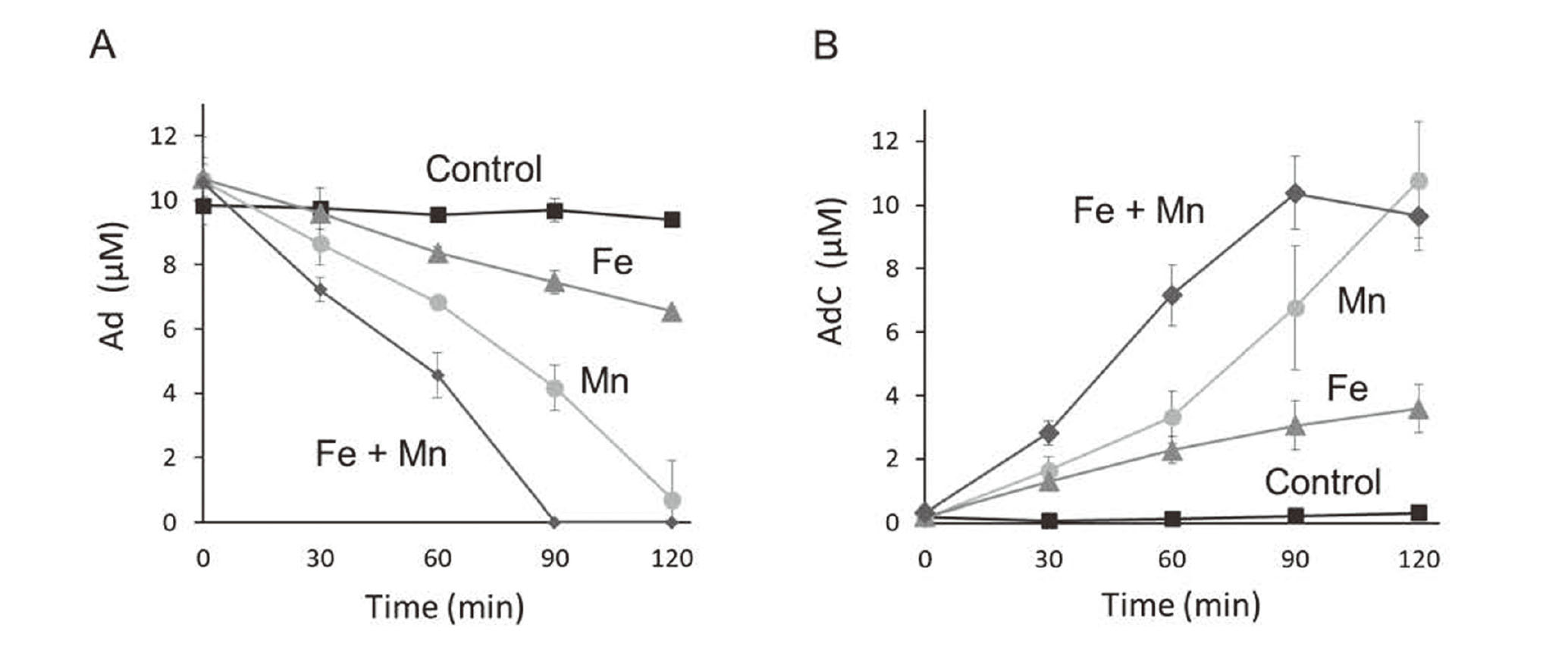
Mn-mediated oxidation of Ad. Ad (10 µM) was mixed with 20 µM Mn(II), 20 µM Fe(III)-NTA, or both and analyzed at the indicated time points by HPLC equipped with a UV-vis detector. The time course of Ad reduction (A) and AdC formation (B) are shown. Data are shown as the mean ± S.D. of three separate experiments.
AdC generates a superoxide anion radical from its autoxidation process (Bindoli et al., 1999). The superoxide anion radical changes into hydrogen peroxide then into a hydroxyl radical by the Fenton reaction. Consistent with these results, formation of 8-oxodG increased in the presence of Fe (Fig. 2A). However, cell viability decreased by the addition of AdC or Ad preincubated with Mn even under conditions where no oxidative DNA damage occurred (Fig. 2B). This suggests there is a cytotoxic mechanism mediated by AdC other than oxidative damage.
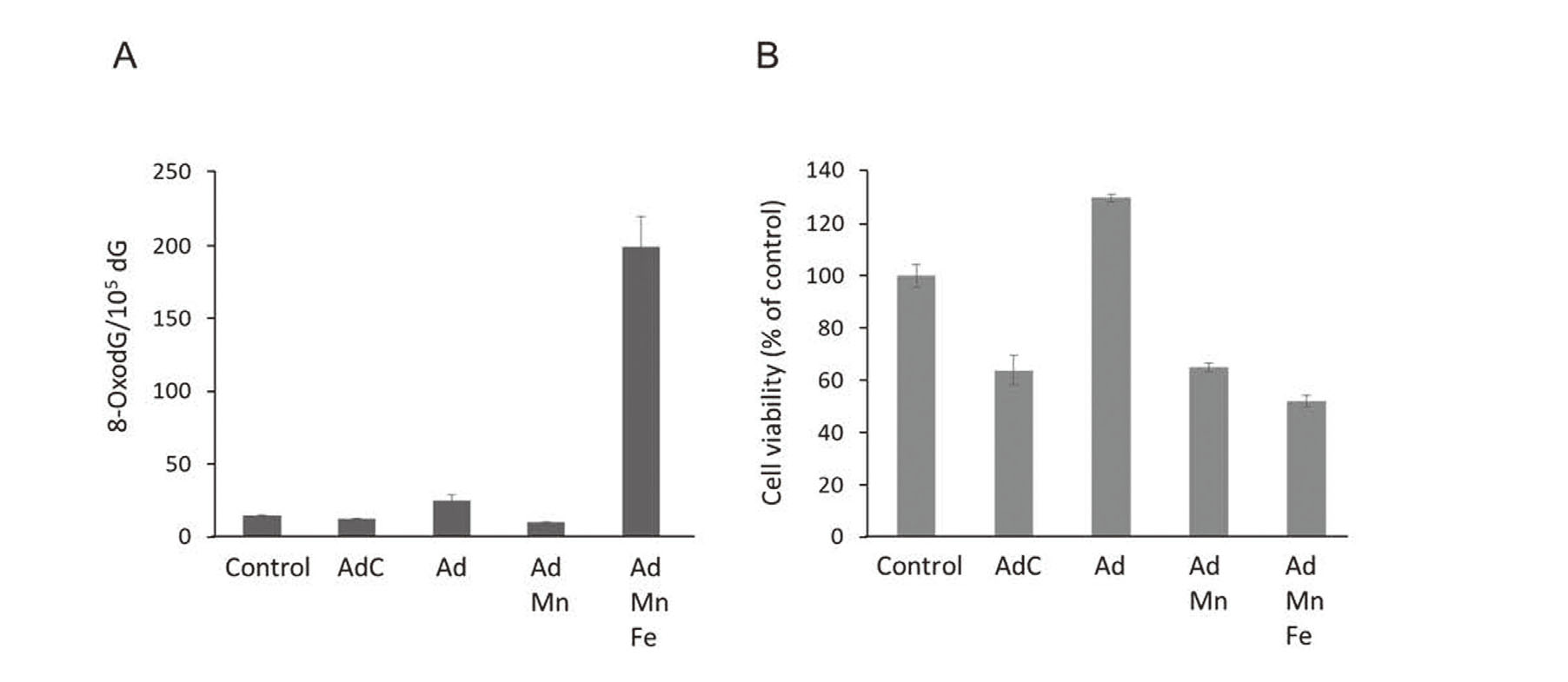
Oxidative DNA damage by Ad, Mn, and Fe as well as cytotoxicity by AdC. Formation of 8-oxodG in salmon sperm DNA exposed for 2 hr to the indicated compounds and metals at concentrations of 10 µM AdC, 10 µM Ad, 1 µM Mn(II), and 20 µM Fe(III)-NTA (A). Viability of SH-SY5Y cells treated for 6 hr with the indicated compounds and metals at concentrations of 20 μM Ad, 40 μM Mn, 20 μM AdC, and 40 μM Fe(III)-NTA (B). Data are shown as the mean ± S.D. of three separate experiments.
Neuronal cell death can be induced by the inhibition of general gene expression (Hoshino et al., 2006; Ishimura et al., 2014), which led us to examine the effect of AdC on gene expression. We applied an in vitro transcription system using bacteriophage T7 RNA polymerase (Nishino et al., 2011; Ueda et al., 2014). The synthesized mRNA was quantified by scanning the resulting agarose gel. There was a slight decrease in mRNA in the Ad-treated system (Fig. 3A). In contrast, AdC decreased mRNA in a dose-dependent manner (Fig. 3B). Ad plus Mn, which produced AdC during incubation, also significantly inhibited transcription (p < 0.05, t-test) in the presence of Mn at 1 µM and greater (Fig. 3C).
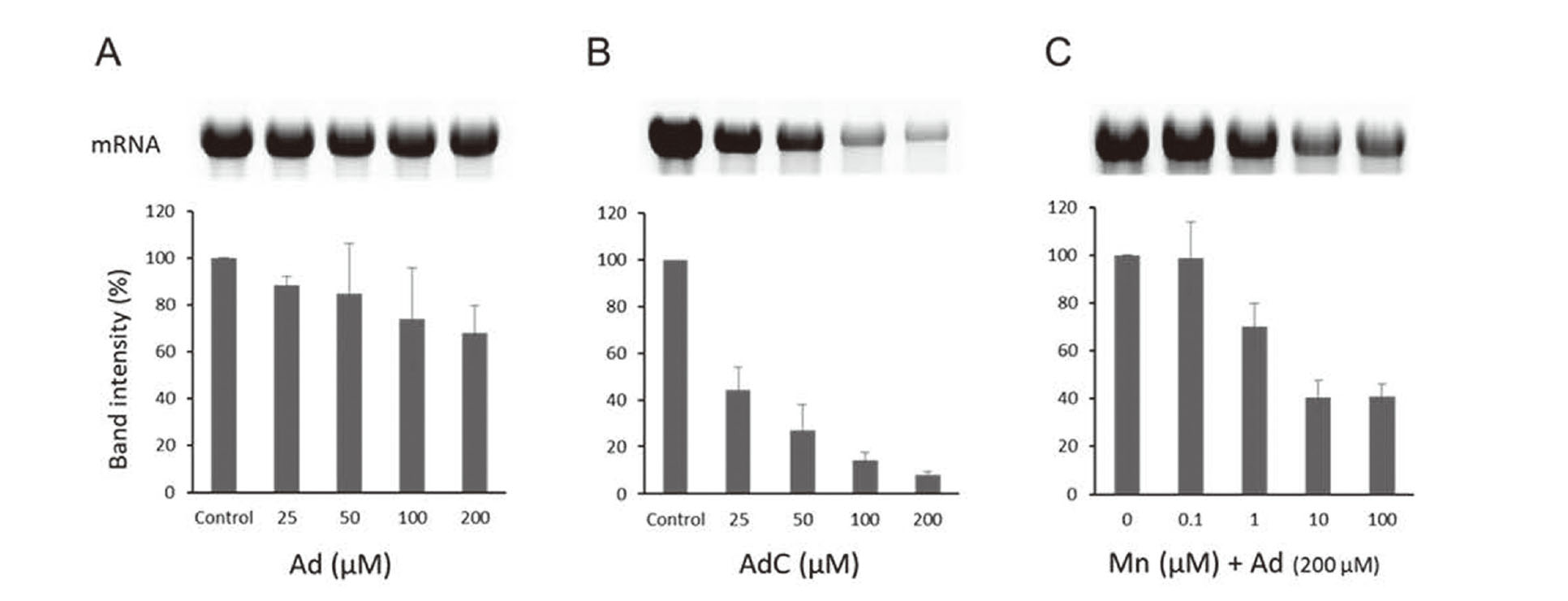
Inhibition of in vitro transcription by AdC. Amount of mRNA synthesized by a system treated with Ad (A), AdC (B), or Mn(II) and Ad (C). T7 RNA polymerase-driven transcription system was used for mRNA synthesis. The transcripts were denatured and detected by agarose electrophoresis. Gel images are representative, and the band intensities are shown as the mean ± S.D. of three separate experiments.
The inhibitory effect of AdC was examined in a human cell line using a nascent RNA detection system based on the incorporation of modified EU followed by fluorescent labeling. The prepared RNA showed increased fluorescence in the presence of EU. The addition of AdC suppressed fluorescence in a dose-dependent manner (Fig. 4). Although the detectable response was different by cell type, the potential of AdC to work against transcription would result in the dysfunction of the neuronal system due to lifelong exposure.
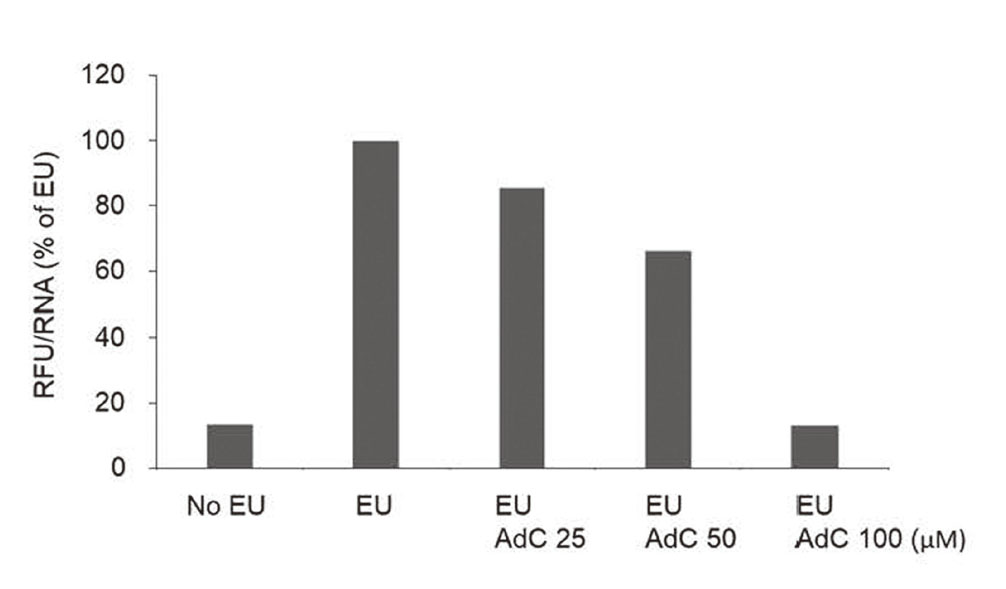
Transcriptional activity of AdC-treated cells. Newly synthesized RNA were labeled with EU and fluorescent dye in MCF-7 cells, and its relative fluorescence unit (RFU) per RNA is represented as percent of EU-treated cells. Representative data of three separate experiments are shown.
Figure 5 shows the mechanism for Mn-induced neurotoxicity mediated by catecholamine, which has been demonstrated with Ad in this study. Mn(III) is produced from relatively inert Mn(II) via the dismutation of superoxide anion radical (O2·-) to hydrogen peroxide (H2O2) during catecholamine autooxidation (Ando et al., 2011). The reactive Mn(III) accelerates the oxidation of catecholamine. The oxidized product, aminochrome, represses global gene expression at the transcriptional level and decreases cell viability. However, in the presence of Fe, Fe(III) is reduced to Fe(II) and catalyzes the generation of a hydroxyl radical (HO·) which induces oxidative DNA damage (Nishino et al., 2011). These mechanisms to disrupt DNA structure (oxidative damage) and function (deficient transcription) may work to induce Mn neurotoxicity.
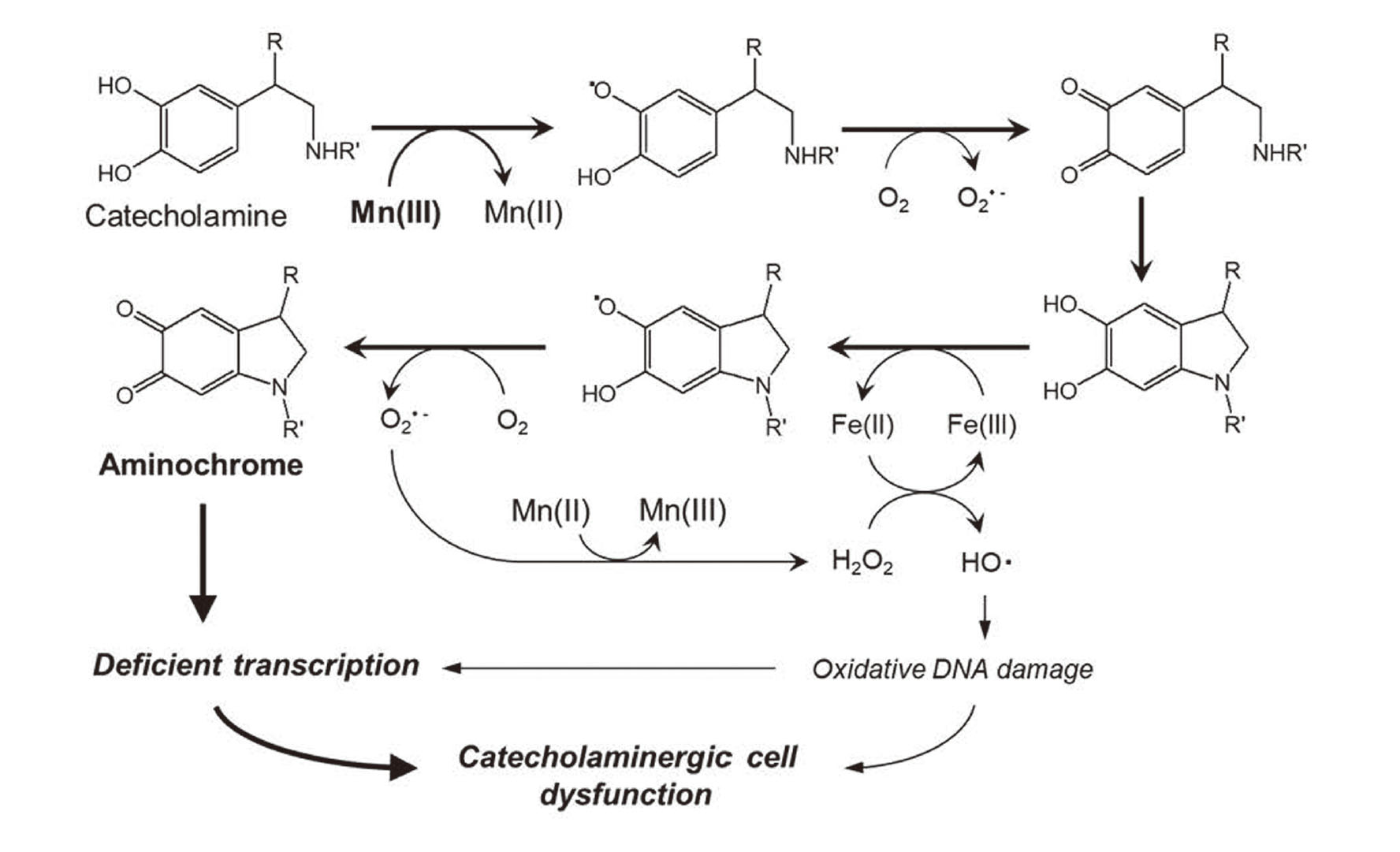
Possible mechanism for Mn neurotoxicity involving aminochrome-mediated transcriptional inhibition.
The authors would like to thank all the laboratory members involved in this work, especially Ms. Misato Inoue for preparing most of the data used in this paper. This study was supported in part by the Academic Research Institute of Meijo University.
Conflict of interestThe authors declare that there is no conflict of interest.