Abstract
Coumarin is a dietary-derived substance that is extensively metabolized by human liver to excretable 7-hydroxycoumarin. Although coumarin under daily dietary consumption is generally regarded as nontoxic, the substance is of toxicological and clinical interest because of its potential association with hepatotoxicity, which is especially evident in rats. In this study, the pharmacokinetics of coumarin were modeled after virtual oral administration in humans. The adjusted monitoring equivalents of coumarin, along with the biotransformation of coumarin to o-hydroxyphenylacetic acid (via 3,4-epoxidation) based on reported plasma concentrations from rat studies, were scaled to human coumarin equivalents using known species allometric scaling factors. Using rat and human liver preparations, data on the rapid in vitro metabolic clearance for humans (~50-fold faster than in rats) were obtained for in vitro–in vivo extrapolation. For human physiologically based pharmacokinetic (PBPK) modeling, the metabolic ratios to o-hydroxyphenylacetic acid and 7-hydroxycoumarin were set at minor (0.1) and major (0.9) levels for the total disappearance of coumarin. The resulting modeled plasma concentration curves in humans generated by simple PBPK models were consistent with reported simulated coumarin maximum concentrations. These results provide basic information to simulate plasma levels of coumarin and its primary metabolite 7-hydroxycoumarin or its secondary activated metabolite o-hydroxyphenylacetic acid (via 3,4-epoxidation) resulting from dietary foodstuff consumption. Under the current assumptions, little toxicological impact of coumarin was evident in humans, thereby indicating the usefulness of forward dosimetry using PBPK modeling for human risk assessment.
INTRODUCTION
Coumarin (Fig. 1A) is a naturally occurring organic chemical found in food products, in various plants, and in essential oils, such as cinnamon and flowering cherry (Lake, 1999; Abraham et al., 2010). Commercially, coumarin is used for its fragrance, and it has been estimated that coumarin is included in 90% of cosmetic and other fragranced products (Born et al., 2000). Coumarin causes hepatotoxicity in rats via the reactive metabolite coumarin 3,4-epoxide (Rietjens et al., 2010; Vassallo et al., 2003). Nonetheless, coumarin has been used as a medicine in humans, with a tolerable daily intake of 0.1 mg/kg body weight (Lake, 1999; Abraham et al., 2010). Heavy consumers of cassia cinnamon would likely reach a daily coumarin intake corresponding to the tolerable daily intake; however, the risk of severe hepatotoxicity in humans is generally estimated to be low, probably because the major metabolic pathway of coumarin in humans is different from that in rats (Rietjens et al., 2008; Vassallo et al., 2004). In in vitro experiments using human liver microsomes, coumarin was rapidly eliminated, mainly via 7-hydroxylation by cytochrome P450 2A6 (Yamazaki et al., 1994) with a minor contribution of 3,4-epoxidation by cytochrome P450 2E1 (Born et al., 2002). In contrast, in experiments with rat liver microsomes, coumarin was slowly eliminated via 3,4-epoxidation forming o-hydroxyphenylacetic acid as a major metabolite (Tanaka et al., 2017). Although clinical data on hepatotoxicity from patients treated with coumarin as a medicinal drug and data on healthy subjects under the consumption of dietary foodstuff are available, there is currently no in-depth report on the in vivo metabolic profiles of coumarin in human plasma.

Full-scale or simplified physiologically based pharmacokinetic (PBPK) modeling has been recommended for interpreting toxicity screening data for general food chemicals including coumarin (Sweeney et al., 2010; Rietjens et al., 2008; Vassallo et al., 2004). To emphasize their utility and simplicity compared with complex full multi-compartment models, we developed a simple modeling system that uses a combination of algorithms with empirical data and literature resources (Kamiya et al., 2019; Miura et al., 2019a, 2019b). In the present study, using known species allometric scaling factors, the pharmacokinetics of coumarin after oral administration were modeled in humans based on the reported plasma concentrations in a rat study in which with main biotransformation of coumarin was to o-hydroxyphenylacetic acid (via 3,4-epoxidation). By applying forward dosimetry after virtual oral administration of coumarin, we report herein the estimated in silico human plasma concentrations of coumarin and 7-hydroxycoumarin (a total of free and glucuronide forms) or o-hydroxyphenylacetic acid.
MATERIALS AND METHODS
Rat liver microsomes from 7-week-old, male Sprague-Dawley rats were prepared previously (Miura et al., 2019b); this animal study was approved by the Ethics Committee of Showa Pharmaceutical University. In vitro elimination rates of coumarin (Fujifilm Wako Pure Chemicals, Osaka, Japan) catalyzed by pooled liver microsomes from rats or humans (H150, Corning, Woburn, MA, USA) were measured using liquid chromatography with fluorescence and ultraviolet detectors with an analytical reversed-phase column (Yamazaki et al., 1999). Briefly, to evaluate the elimination of coumarin, substrate (1.0 µM, i.e., within the linear range) was incubated with liver microsomes (0.50 mg protein/mL) and an NADPH-generating system in a total of 0.50 mL of 100 mM potassium phosphate buffer (pH 7.4) at 37°C for 15 min. In vitro hepatic intrinsic clearance (CLh,int) values were calculated from coumarin elimination rates, with the liver microsomal unbound fraction for coumarin (0.907) determined by Simcyp software, by extrapolation using the following values: 40 mg liver microsomal protein per 1 g liver and 10 g liver weight per 0.25 kg of rat bodyweight or 1.5 kg liver weight per 70 kg of human bodyweight (Miura et al., 2019b; Takano et al., 2010).
Simple PBPK models for coumarin and o-hydroxyphenylacetic acid or 7-hydroxycoumarin consisting of receptor (gut), metabolizing (liver), and central compartments were set up as previously described (Kamiya et al., 2019; Miura et al., 2019a, 2019b). The detailed procedure for establishing a PBPK model was recently described (Kamiya et al., 2019, 2020a). The hepatic blood flow rates (Qh) in rats (0.853 L/hr) and humans (96.6 L/hr) were taken from the literature (Kato et al., 2008). The plasma unbound fraction (fu,p), octanol-water partition coefficient (logP), blood-to-plasma concentration ratio (Rb), and the liver-to-plasma concentration ratio (Kp,h) of the relevant compounds were estimated (Uchimura et al., 2010; Takano et al., 2010; Yamazaki et al., 2016; Kamiya et al., 2019).
The initial values for PBPK modeling for the fraction absorbed × intestinal availability (Fa·Fg), the hepatic clearance (CLh), and the renal clearance (CLr) were derived from the elimination constants in empirical one-compartment models. The Fa value for coumarin of 0.925 was estimated from the apparent permeability across an in vitro intestinal epithelial cell monolayer system (806 nm/sec), as described previously (Kamiya et al., 2020b). Because of limited urinary excretion data, the general ratios of CLh to CLr were set at 9:1 for the substrate and at 5:5 for metabolites in rats. Fg values were estimated from the gut extraction ratios as one-tenth of the hepatic extraction ratios (Eh) in the hepatic well-stirred model (Fg = 1 – Eh/10). To determine the concentrations in each compartment of our PBPK model, the system of differential equations for the compounds under investigation was then solved in a similar way to that carried out previously (Kamiya et al., 2019).
The concentrations of coumarin in plasma from orally treated rats were taken from the literature (Tanaka et al., 2017). The absorption rate constant (ka), volume of the systemic circulation (V1), and hepatic intrinsic clearance (CLh,int) values with standard deviations were determined by fitting using nonlinear regression analyses. The final parameter values for coumarin and o-hydroxyphenylacetic acid or 7-hydroxycoumarin in rat PBPK models are shown in Tables 1 and 2. The resulting system of differential equations was solved for concentrations of the substrate and its metabolites, o-hydroxyphenylacetic acid or 7-hydroxycoumarin (indicated with subscript m):
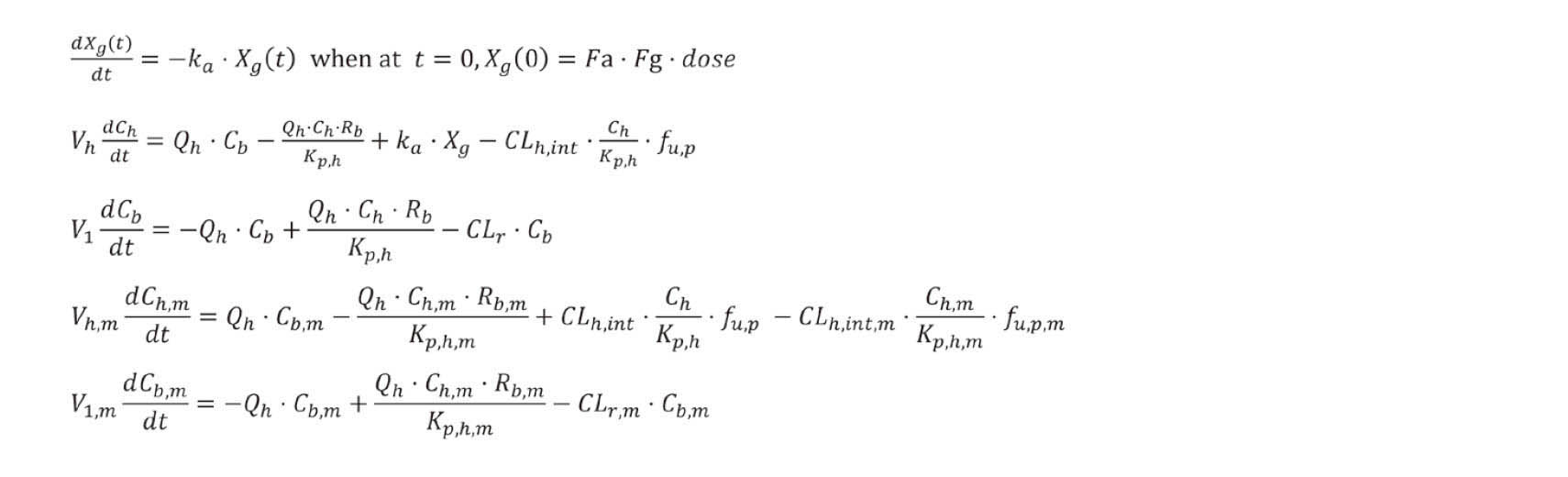
where Ch is the hepatic substrate concentration, Cb is the blood substrate concentration, Vh is the volume of liver, and Xg is the amount of compound in the gut.
Table 1. Chemical properties of coumarin and its metabolites used for PBPK modeling.
|
Coumarin |
o-Hydroxyphenylacetic acid |
7-Hydroxycoumarin |
| Molecular weight |
146 |
152 |
162 |
| Octanol–water partition coefficient, (clogP) |
1.82 (1.41) |
1.15 (0.697) |
1.44 (1.62) |
| Plasma unbound fraction |
0.412 |
0.176 |
0.170 |
| Blood–plasma concentration ratio |
0.923 |
0.881 |
0.878 |
| Liver–plasma concentration ratio |
0.875 |
0.504 |
0.876 |
Table 2. Physiological, experimental, and final calculated parameters for rat and human PBPK models for coumarin,
o-hydroxyphenylacetic acid, and 7-hydroxycoumarin.
| Parameter |
Abbreviation (unit) |
Rat |
Human |
| Fraction absorbed × intestinal availability |
Fa·Fg |
0.973 |
0.837 |
| Absorption rate constant |
ka (1/hr) |
5.06 ± 0.09 a |
3.76 |
| Volume of systemic circulation for coumarin |
V1_substrate (L) |
0.866 ± 0.005 a |
245 |
| Hepatic intrinsic clearance for coumarin |
CLh,int_substrate (L/hr) |
0.746 ± 0.005 a |
5150 |
| Hepatic clearance for coumarin |
CLh,_substrate (L/hr) |
0.226 |
92.4 |
| Renal clearance for coumarin |
CLr,_substrate (L/hr) |
0.023 |
0.967 |
| Volume of systemic circulation for o-hydroxyphenylacetic acid |
V1_o-hydroxyphenylacetic acid (L) |
0.728 ± 0.001 a |
205 |
| Volume of systemic circulation for 7-hydroxycoumarin |
V1_7-hydroxycoumarin (L) |
0.961 ± 0.008 a |
272 |
| Hepatic intrinsic clearance for o-hydroxyphenylacetic acid |
CLh,int_ o-hydroxyphenylacetic acid (L/hr) |
1.64 ± 0.01 a |
11300 |
| Hepatic intrinsic clearance for 7-hydroxycoumarin |
CLh,int_7-hydroxycoumarin (L/hr) |
1.79 ± 0.02 a |
12400 |
| Hepatic clearance for o-hydroxyphenylacetic acid |
CLh_o-hydroxyphenylacetic acid (L/hr) |
0.216 |
92.1 |
| Hepatic clearance for 7-hydroxycoumarin |
CLh_7-hydroxycoumarin (L/hr) |
0.224 |
92.4 |
| Renal clearance for o-hydroxyphenylacetic acid |
CLr_o-hydroxyphenylacetic acid (L/hr) |
0.152 |
6.52 |
| Renal clearance for 7-hydroxycoumarin |
CLr_7-hydroxycoumarin (L/hr) |
0.157 |
6.73 |
a Data are means ± standard deviations. The metabolic ratios to o-hydroxyphenylacetic acid and 7-hydroxycoumarin were 0.9 and 0.0025 for rats, respectively, and 0.1 and 0.9 for humans. Parameters such as Kp,h, Rb, and fu,p were assumed to be the same for humans and for rats.
To set up the human PBPK models of coumarin and o-hydroxyphenylacetic acid or 7-hydroxycoumarin based on the rat PBPK model parameters, liver microsomal elimination rates and physiological parameters for humans derived from the literature were used. The values of ka, V1, and CLh for coumarin were determined by a scale-up strategy from rats to humans (Shimizu and Yamazaki, 2017; Miura et al., 2019b). The absorption rate constant (ka) in rats was multiplied by 0.744 to give the human ka value. The human systemic circulation volume (V1,human) was estimated using Vh and the blood volume (Vb), with Vh,human, Vb,rat, and Vb,human values of 1.50, 0.0160, and 4.90 L, respectively (Shimizu and Yamazaki, 2017; Miura et al., 2019b):

where Fh is the unmetabolized fraction in liver, Vss is the volu me of distribution at steady state, and the value of 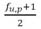 is corresponded to fu,p to tissue fraction unbound. The in vivo hepatic intrinsic clearance (CLh,int) in humans was estimated by multiplying the calculated initial parameters for in vitro hepatic intrinsic clearance in humans by the ratio of in vivo to in vitro hepatic intrinsic clearance in rats, as mentioned above for modeling in rats (Takano et al., 2010; Miura et al., 2019b; Shimizu and Yamazaki, 2017) The human renal clearance CLr,human was estimated using the equation, where BWrodent = 0.25 kg (rat) and BWhuman = 70 kg:
is corresponded to fu,p to tissue fraction unbound. The in vivo hepatic intrinsic clearance (CLh,int) in humans was estimated by multiplying the calculated initial parameters for in vitro hepatic intrinsic clearance in humans by the ratio of in vivo to in vitro hepatic intrinsic clearance in rats, as mentioned above for modeling in rats (Takano et al., 2010; Miura et al., 2019b; Shimizu and Yamazaki, 2017) The human renal clearance CLr,human was estimated using the equation, where BWrodent = 0.25 kg (rat) and BWhuman = 70 kg:

The final parameter values for humans are shown in Table 2. The above-described system of differential equations was also solved to determine the concentrations in each compartment in humans. The modeled human plasma concentration curves of coumarin were verified in comparison with the reported estimated curves (Ritschel et al., 1977).
RESULTS AND DISCUSSION
Under our extensive literature survey, only in vivo pharmacokinetic data points for the foodstuff coumarin were only available in rats. Figure 1A illustrates the reported pathways of coumarin and its metabolites (Born et al., 2000) as detected in plasma from rats after oral administration of 200 mg/kg (Tanaka et al., 2017). Figure 1B shows the reported levels of coumarin and its metabolites in rat plasma after oral administration of coumarin (Tanaka et al., 2017). From these concentrations, the values of ka, V1, and CLh,int for use in the rat PBPK model were determined by fitting procedures and are shown in Table 2. The metabolic ratios of coumarin to o-hydroxyphenylacetic acid and 7-hydroxycoumarin were set at 0.9 and 0.0025 for rats, respectively, in accordance with the plasma concentration curves in rats. By solving the equations that make up the PBPK model, virtual plasma concentration curves were generated for rats, and the resulting in silico concentration curves are shown in Fig. 1B. These PBPK-generated lines were consistent with the reported in vivo data points.
To set up human PBPK models for coumarin and its metabolites, the in vitro substrate elimination of coumarin was investigated using liver microsomes from rats and humans. Under the present conditions, the metabolic elimination rate of coumarin by human liver microsomes (2.3 nmol/min/mg protein) was 46-fold faster than that by rat liver microsomes (0.05 nmol/min/mg protein). Parameters for the human PBPK model for coumarin (Table 2) were established based on the parameters in the rat PBPK model. In silico plasma concentration curves after the virtual administration of coumarin in humans were created using these PBPK parameters. Figure 1C shows in silico concentration curves for coumarin and its metabolites after oral administration of 60 mg in humans (70 kg body weight); these curves are consistent with the reported simulated coumarin pharmacokinetics in humans (i.e., a maximum concentration of 0.01 µg/mL of coumarin with extensive 7-hydroxylations) generated using a two-compartment model (Ritschel et al., 1977).
The difference should be noted between the reported rapid elimination of coumarin via 7-hydroxylation in humans (Peamkrasatam et al., 2006; Ujjin et al., 2002; Kiyotani et al., 2003) compared with rats, in which the 7-hydroxylation of coumarin is slow (Yamazaki et al., 1994) so the activation to reactive 3,4-epoxide is more important (Fig. 1A). In a preliminary study with 9000 × g supernatant fractions from rat and human livers, we found roughly similar rates of biotransformation of coumarin to o-hydroxyphenylacetic acid (2-4 pmol/min/mg protein), but slow and rapid 7-hydroxylation activities, respectively, of rat and human livers (5 versus 90 pmol/min/mg protein). The metabolic ratios for o-hydroxyphenylacetic acid and 7-hydroxycoumarin were set to minor (0.1) and major (0.9) levels, respectively, for the total coumarin disappearance in humans under the present PBPK modeling conditions, and the resulting concentration curves are shown in Fig. 1C.
The present PBPK models succeeded in estimating human plasma concentrations of coumarin and its metabolites, o-hydroxyphenylacetic acid and 7-hydroxycoumarin (a total of free and glucuronide forms), after ingestion of coumarin; this forward dosimetry approach could be applied to toxicological risk assessments. To further develop toxicological assessments in humans, the current PBPK model could also be used to assess the genetic influences of cytochrome P450 2A6 variations (Peamkrasatam et al., 2006; Ujjin et al., 2002; Kiyotani et al., 2003) on the human CLh,int values in in vivo coumarin pharmacokinetics. Further investigations with human hepatocytes in vitro or with human hepatocytes transplanted into an immunodeficient mouse model would be of interest. Such approaches will support further characterization of the metabolic clearance rates and balances of these activated/deactivated metabolites from food-derived coumarin mediated by cytochromes P450 and will foster further improvement of the current human PBPK models.
In conclusion, in the present study, the metabolic profiles of coumarin, o-hydroxyphenylacetic acid, and 7-hydroxycoumarin in human plasma were generated by simple PBPK modeling after virtual oral doses of coumarin. The human plasma levels and the ratios of coumarin and 7-hydroxycoumarin will undoubtedly be affected by interindividual variability. However, under the current assumptions, the metabolic ratios of coumarin to o-hydroxyphenylacetic acid, generated via 3,4-epoxidation, indicate that this is a minor pathway; nonetheless, it is evident that this metabolic activation process does exist in humans. Further studies will be needed to investigate whether cytochrome P450 2A6 poor metabolizers are at risk from o-hydroxyphenylacetic acid-induced hepatotoxicity. The current results suggested that the forward or reverse dosimetry of coumarin and o-hydroxyphenylacetic acid or 7-hydroxycoumarin using the newly established simple PBPK model could contribute to human risk assessment of dietary-derived coumarin.
ACKNOWLEDGMENTS
The authors thank Drs. Takashi Yamada, Yasuhiro Tanaka, and Shohei Otsuka for their assistance and David Smallbones for copyediting a draft of this article. This work was supported in part by the Food Safety Commission of Japan (JPCAFSC20202006) and the METI Artificial Intelligence-based Substance Hazard Integrated Prediction System Project, Japan. TM and YK, respectively, were partly supported by the Japan Society for the Promotion of Science Grant-in-Aid for Young Scientists 202021210 and 19K16422.
Conflict of interest
The authors declare that there is no conflict of interest.
REFERENCES
- Abraham, K., Wöhrlin, F., Lindtner, O., Heinemeyer, G. and Lampen, A. (2010): Toxicology and risk assessment of coumarin: focus on human data. Mol. Nutr. Food Res., 54, 228-239.
- Born, S.L., Caudill, D., Fliter, K.L. and Purdon, M.P. (2002): Identification of the cytochromes P450 that catalyze coumarin 3,4-epoxidation and 3-hydroxylation. Drug Metab. Dispos., 30, 483-487.
- Born, S.L., Caudill, D., Smith, B.J. and Lehman-McKeeman, L.D. (2000): In vitro kinetics of coumarin 3,4-epoxidation: application to species differences in toxicity and carcinogenicity. Toxicol. Sci., 58, 23-31.
- Kamiya, Y., Otsuka, S., Miura, T., Takaku, H., Yamada, R., Nakazato, M., Nakamura, H., Mizuno, S., Shono, F., Funatsu, K. and Yamazaki, H. (2019): Plasma and hepatic concentrations of chemicals after virtual oral administrations extrapolated using rat plasma data and simple physiologically based pharmacokinetic models. Chem. Res. Toxicol., 32, 211-218.
- Kamiya, Y., Otsuka, S., Miura, T., Yoshizawa, M., Nakano, A., Iwasaki, M., Kobayashi, Y., Shimizu, M., Kitajima, M., Shono, F., Funatsu, K. and Yamazaki, H. (2020a): Physiologically based pharmacokinetic models predicting renal and hepatic concentrations of industrial chemicals after virtual oral doses in rats. Chem. Res. Toxicol., 33, 1736-1751.
- Kamiya, Y., Takaku, H., Yamada, R., Akase, C., Abe, Y., Sekiguchi, Y., Murayama, N., Shimizu, M., Kitajima, M., Shono, F., Funatsu, K. and Yamazaki, H. (2020b): Determination and prediction of permeability across intestinal epithelial cell monolayer of a diverse range of industrial chemicals/drugs for estimation of oral absorption as a putative marker of hepatotoxicity. Toxicol. Rep., 7, 149-154.
- Kato, M., Shitara, Y., Sato, H., Yoshisue, K., Hirano, M., Ikeda, T. and Sugiyama, Y. (2008): The quantitative prediction of CYP-mediated drug interaction by physiologically based pharmacokinetic modeling. Pharm. Res., 25, 1891-1901.
- Kiyotani, K., Yamazaki, H., Fujieda, M., Iwano, S., Matsumura, K., Satarug, S., Ujjin, P., Shimada, T., Guengerich, F.P., Parkinson, A., Honda, G., Nakagawa, K., Ishizaki, T. and Kamataki, T. (2003): Decreased coumarin 7-hydroxylase activities and CYP2A6 expression levels in humans caused by genetic polymorphism in CYP2A6 promoter region (CYP2A6*9). Pharmacogenetics, 13, 689-695.
- Lake, B.G. (1999): Coumarin metabolism, toxicity and carcinogenicity: relevance for human risk assessment. Food Chem. Toxicol., 37, 423-453.
- Miura, T., Uehara, S., Mizuno, S., Yoshizawa, M., Murayama, N., Kamiya, Y., Shimizu, M., Suemizu, H. and Yamazaki, H. (2019a): Steady-state human pharmacokinetics of monobutyl phthalate predicted by physiologically based pharmacokinetic modeling using single-dose data from humanized-liver mice orally administered with dibutyl phthalate. Chem. Res. Toxicol., 32, 333-340.
- Miura, T., Uehara, S., Nakazato, M., Kusama, T., Toda, A., Kamiya, Y., Murayama, N., Shimizu, M., Suemizu, H. and Yamazaki, H. (2019b): Human plasma and liver concentrations of styrene estimated by combining a simple physiologically based pharmacokinetic model with rodent data. J. Toxicol. Sci., 44, 543-548.
- Peamkrasatam, S., Sriwatanakul, K., Kiyotani, K., Fujieda, M., Yamazaki, H., Kamataki, T. and Yoovathaworn, K. (2006): In vivo evaluation of coumarin and nicotine as probe drugs to predict the metabolic capacity of CYP2A6 due to genetic polymorphism in Thais. Drug Metab. Pharmacokinet., 21, 475-484.
- Rietjens, I.M., Boersma, M.G., Zaleska, M. and Punt, A. (2008): Differences in simulated liver concentrations of toxic coumarin metabolites in rats and different human populations evaluated through physiologically based biokinetic (PBBK) modeling. Toxicol. In Vitro, 22, 1890-1901.
- Rietjens, I.M., Punt, A., Schilter, B., Scholz, G., Delatour, T. and van Bladeren, P.J. (2010): In silico methods for physiologically based biokinetic models describing bioactivation and detoxification of coumarin and estragole: implications for risk assessment. Mol. Nutr. Food Res., 54, 195-207.
- Ritschel, W.A., Brady, M.E., Tan, H.S., Hoffmann, K.A., Yiu, I.M. and Grummich, K.W. (1977): Pharmacokinetics of coumarin and its 7-hydroxy-metabolites upon intravenous and peroral administration of coumarin in man. Eur. J. Clin. Pharmacol., 12, 457-461.
- Shimizu, M. and Yamazaki, H. (2017): Human plasma and urinary metabolic profiles of trimethylamine and trimethylamine N-oxide extrapolated using a simple physiologically based pharmacokinetic model. J. Toxicol. Sci., 42, 485-490.
- Sweeney, L.M., Kirman, C.R., Gargas, M.L., Carson, M.L. and Tardiff, R.G. (2010): Development of a physiologically-based toxicokinetic model of acrylamide and glycidamide in rats and humans. Food Chem. Toxicol., 48, 668-685.
- Takano, R., Murayama, N., Horiuchi, K., Kitajima, M., Kumamoto, M., Shono, F. and Yamazaki, H. (2010): Blood concentrations of acrylonitrile in humans after oral administration extrapolated from in vivo rat pharmacokinetics, in vitro human metabolism, and physiologically based pharmacokinetic modeling. Regul. Toxicol. Pharmacol., 58, 252-258.
- Tanaka, Y., Fujii, W., Hori, H., Kitagawa, Y. and Ozaki, K. (2017): Changes in coumarin kinetics and subcellular localization of CYP2E1 contribute to bile duct damage and reduce hepatocellular damage after repeated administration of coumarin in rats. Toxicol. Lett., 280, 99-105.
- Uchimura, T., Kato, M., Saito, T. and Kinoshita, H. (2010): Prediction of human blood-to-plasma drug concentration ratio. Biopharm. Drug Dispos., 31, 286-297.
- Ujjin, P., Satarug, S., Vanavanitkun, Y., Daigo, S., Ariyoshi, N., Yamazaki, H., Reilly, P.E., Moore, M.R. and Kamataki, T. (2002): Variation in coumarin 7-hydroxylase activity associated with genetic polymorphism of cytochrome P450 2A6 and the body status of iron stores in adult Thai males and females. Pharmacogenetics, 12, 241-249.
- Vassallo, J.D., Hicks, S.M., Daston, G.P. and Lehman-McKeeman, L.D. (2004): Metabolic detoxification determines species differences in coumarin-induced hepatotoxicity. Toxicol. Sci., 80, 249-257.
- Vassallo, J.D., Morrall, S.W., Fliter, K.L., Curry, S.M., Daston, G.P. and Lehman-McKeeman, L.D. (2003): Liquid chromatographic determination of the glutathione conjugate and ring-opened metabolites formed from coumarin epoxidation. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci., 794, 257-271.
- Yamazaki, H., Mimura, M., Sugahara, C. and Shimada, T. (1994): Catalytic roles of rat and human cytochrome P450 2A enzymes in testosterone 7 alpha- and coumarin 7-hydroxylations. Biochem. Pharmacol., 48, 1524-1527.
- Yamazaki, H., Suemizu, H., Mitsui, M., Shimizu, M. and Guengerich, F.P. (2016): Combining chimeric mice with humanized liver, mass spectrometry, and physiologically-based pharmacokinetic modeling in toxicology. Chem. Res. Toxicol., 29, 1903-1911.
- Yamazaki, H., Tanaka, M. and Shimada, T. (1999): Highly sensitive high-performance liquid chromatographic assay for coumarin 7-hydroxylation and 7-ethoxycoumarin O-deethylation by human liver cytochrome P450 enzymes. J. Chromatogr. B Biomed. Sci. Appl., 721, 13-19.