Abstract
Acrylonitrile (AN), which is widely utilized in the manufacture of plastics, acrylamide, acrylic fibers, and resins, is also one of main components of cigarette smoke (CS). In this study, we examined the effects of AN on the cell viability and apoptosis of JEG-3 and BeWo human choriocarcinoma cancer cell lines. A cell viability assay confirmed that AN decreased the cell proliferation of JEG-3 and BeWo cells in a dose-dependent manner. Additionally, Western blot assay revealed that protein expression of cyclin D and cyclin E decreased, while protein expression of p21 and p27 increased in response to AN treatment for 48 hr. The changes in reactive oxygen species (ROS) levels in JEG-3 and BeWo cells exposed to AN were also measured by a dichlorofluorescein diacetate (DCFH-DA) assay, which revealed that ROS levels increased in response to AN treatment for 48 hr. Moreover, western blot assay confirmed that AN treatment of JEG-3 and BeWo cells for 4 hr promoted the expression of phosphorylated eukaryotic initiation factor 2 alpha protein (p-eIF2α), C/EBP homologous protein (CHOP) and caspase 12, which are known to be involved in ROS-mediated endoplasmic reticulum stress (ER-stress)-related apoptosis. Overall, the protein expression of p53 and Bax (a pro-apoptosis marker) increased, while the expression of Bcl-xl (an anti-apoptotic marker) decreased and the number of apoptotic cells increased in response to AN treatment for 48 hr. Taken together, these results suggest that AN has the potential to induce apoptosis of JEG-3 and BeWo human choriocarcinoma cancer cells by activating ROS.
INTRODUCTION
Acrylonitrile (AN) is widely utilized in the manufacture of plastics, acrylamide, acrylic fibers, synthetic rubber, resins, and other chemicals. Humans are usually exposed to AN through air, food, drinking water, and cigarette smoke (CS). According to the WHO, the main exposure pathway to AN is occupational exposure, while cigarette smoking may be the most important source of non-occupational AN exposure (World Health Organization, 2000). Indeed, AN is one of the main components of CS (Nazaroff and Singer, 2004), and mainstream CS discharged from one cigarette reportedly contains 7.8 to 39.1 μg of AN (Haber and Patterson, 2005). In addition, CS appears to be one of the hazardous factors responsible for various cancers via a variety of mechanisms (Sobus and Warren, 2014). In recent years, the rate of female smoking has been significantly increasing in the world, and the proportion of women at risk of developing diseases caused by CS is also increasing (Duan et al., 2017; Satcher et al., 2002; Jeon et al., 2016). Since AN has been ranked as a major ingredient of CS and recently women’s smoking rates have been significantly increasing, the outcomes of this study will also help elucidate the correlation of cytotoxicity of AN derived from CS with the placental function of pregnant women.
AN is severely toxic to humans and is a Class 2B carcinogen according to the International Agency for Research on Cancer (IARC) (IARC, 1999). In a previous study, a significant increase in lung cancer was reported in workers chronically exposed to AN (Sponsiello-Wang et al., 2006). The occurrence of primary brain tumors has been also found to be associated with AN (Kirman et al., 2005), and a major target of AN is known to be the central nervous system (Yu et al., 2016). In primary astrocytes of the brain exposed to AN, gliosis occurred via increased formation of reactive oxygen species (ROS), ATP depletion, and DNA damage, which were induced by AN (Enongene et al., 2000; Esmat et al., 2007). Moreover, a previous study reported that AN inhibited gap junctional intercellular communication through an oxidative stress mechanism in a rat astrocyte cell line (Kamendulis et al., 1999). The causative relationship between AN exposure and adverse outcomes in the nervous system has also been reported (Khan et al., 2009; Yu et al., 2016). Although it is known that oxidative stress plays a role in AN-mediated toxicity, the underlying mechanisms of AN toxicity have yet to be elucidated.
Oxidative stress is triggered by the increased formation of ROS such as oxygen free radicals and peroxides because of oxygen imbalance in the body. Reactive oxygen species cause damage to biological components through oxidation of proteins, lipids and DNA. Thus, oxidative stress can be an obstacle to normal mechanisms of cellular signalings and trigger cell death. Moreover, ROS are considered the cause of aging, immunocompromised conditions and various diseases, as well as one of the key factors determining the toxicity toward regulatory pathways (Kitamura and Hiramatsu, 2010).
Generally, ROS are considered essential for the regulation of normal physiological functions associated with cell cycle progression, proliferation, differentiation, migration and cell death (Carmody et al., 2016; Ray et al., 2012; Redza-Dutordoir and Averill-Bates, 2016). Cell cycle control is mediated by cyclins and cyclin-dependent kinases (CDKs), and ROS have been found to influence cell cycle progression via phosphorylation and ubiquitination of CDKs and cell cycle-related proteins (Verbon et al., 2012). In addition, oxidative stress by ROS can cause damage to cellular molecules and organelles, leading to activation of apoptosis, by regulating the main apoptotic pathways mediated by mitochondria, the endoplasmic reticulum (ER), etc. (Redza-Dutordoir and Averill-Bates, 2016). On the other hand, it has been known that ROS have also anti-apoptotic effects: in some systems, nitric oxide inhibited TNF- or Fas ligand-mediated apoptosis (Simon et al., 2000; Shen et al., 1998; Hebestreit et al., 1998). The diverse or conflicting effects of ROS primarily depend on their concentrations in the cell; however, whether the consequences resulting from the specific ROS effect are harmful or beneficial depends on the cellular conditions.
Globally, the number of women who smoke cigarettes or are exposed to CS through secondhand smoking has been increasing. Therefore, CS now has a greater chance of influencing the health of women, including those who are pregnant. By investigating the cytotoxic effect of AN which is one of main components of CS on choriocarcinoma cell lines that develop only in pregnant women, we can also infer the toxicity of CS to placental function of pregnant women. Thus, in this study, the effects of AN on the growth and apoptosis of two human choriocarcinoma cell lines, JEG-3 and BeWo, were investigated to determine the in vitro toxicity of AN against human placenta-derived cells. Both cell lines have long been studied to investigate placental toxicity-related mechanisms of various compounds (Zhang et al., 1995). Specifically, AN was evaluated for its capacity to induce oxidative stress via ROS formation in JEG-3 and BeWo human choriocarcinoma cancer cell lines, while focusing on its ability to induce apoptosis of these cells by increasing endoplasmic reticulum (ER) stress through ROS formation.
MATERIALS AND METHODS
Reagents and chemicals
Acrylonitrile (AN) was purchased from Sigma-Aldrich Corp. (St. Louis, MO, USA). All chemicals were dissolved in 100% dimethyl sulfoxide (DMSO; Junsei Chemical Co., Tokyo, Japan) to a concentration of 1 M prior to use.
Cell culture and media
The human placenta choriocarcinoma cell lines, JEG-3 and BeWo, were purchased from the Korean Cell Line Bank (Seoul, Korea). JEG-3 cells were cultured using DMEM (HyClone Laboratories, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS; RMBIO, Rocky Mountain Biologicals, Missoula, MT, USA), 1% penicillin G and streptomycin (Cellgro; Mediatech, Corning, NY, USA), and 1% HEPES (Invitrogen Life Technologies, Carlsbad, CA, USA) at 37°C in a humidified atmosphere containing 5% CO2. BeWo cells were cultured using DMEM supplemented with 15% FBS, 1% penicillin G and streptomycin, and 1% HEPES at 37°C in a humidified atmosphere containing 5% CO2. The JEG-3 and BeWo cells were subcultured twice a week. The media change was performed every 48 hr and cells usually were passaged again once an 85~95% confluence was reached.
Cell viability assay
JEG-3 and BeWo cells were seeded at 3 × 103 per well in 96-well plates (SPL Life Science) in a humidified atmosphere of 5% CO2 at 37°C. After the cells were incubated with DMEM medium for 24 hr, they were treated with AN at 10-9 to 10-3 M in DMEM supplemented with 0.1% DMSO for 6 days. During this period, the media were replaced with new media every third day. DMSO was used as a vehicle to carry the chemicals to the media.
Cell viability was determined using a EZ-Cytox cell viability assay kit (iTSBiO, Seoul, Korea). Briefly, EZ-Cytox Kit reagent was added into each well for 2 hr under standard culture conditions, after which 96-well plates were gently shaken thoroughly for 5 min on a rocker at room temperature (RT). The absorbance of the treated and untreated samples at 450 nm was then measured on a multi-well microplate reader (VERSA man). DMEM medium supplemented with the same volume of kit reagent on an empty well was used as a blank. Cell viability was represented by percentage values compared to a control and then used to calculate the number of viable cells.
Protein extraction and Western blot assay
After treatment of JEG-3 and BeWo cells with AN at 10-8 M and 10-6 M, whole cell lysates of JEG-3 and BeWo cells were prepared in 80 μL 1 × RIPA buffer (50 mM Tris-HCl; pH 8, 150 mM NaCl, 1% Triton X-100 (Sigma-Aldrich)), 0.5% deoxycholic acid (Sigma-Aldrich), and 0.1% SDS. Total protein concentrations were quantified using bicinchoninic acid (BCA; Sigma-Aldrich), after which 50 μg of total protein was separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were then transferred to polyvinylidene difluoride (PVDF) membranes (BioRad Laboratories, Hercules, CA, USA), after which the membranes were blocked by treatment with 5% bovine serum albumin (BSA; Sigma-Aldrich) for 120 min at room temperature. The membrane was then incubated overnight at 4°C with mouse monoclonal anti-GAPDH antibody (1:12,000 dilution: Abcam, Cambridge, UK), mouse monoclonal anti-cyclin D antibody (1:3000 dilution; Abcam), rabbit polyclonal anti-cyclin E antibody (1:1000 dilution; Abcam), mouse monoclonal anti-p21 antibody (1:1000 dilution; Abcam), rabbit monoclonal anti-p27 antibody (1:1000 dilution; Abcam), mouse monoclonal anti-Bax (1:1000 dilution; Santa Cruz Biotechnology, Dallas, TX, USA), rabbit monoclonal anti-Bcl-xl (1:1000 dilution; Cell Signaling Technology, Danvers, MA, USA), mouse monoclonal anti-CHOP (1:1000 dilution; Cell Signaling Technology), rabbit monoclonal anti-Nrf2 (1:2000 dilution; Abcam), and rabbit polyclonal anti-phospho-eIF2α antibody (1:1000 dilution; Cell Signaling Technology), mouse monoclonal anti-caspase 3 (1:1000 dilution; Santa Cruz Biotechnology), mouse monoclonal anti-caspase 12 (1:1000 dilution; Santa Cruz Biotechnology). Primary antibody binding was detected with horse radish peroxidase (HRP)-conjugated anti-mouse IgG or anti-rabbit IgG (1:2000, Thermo Scientific) treatment for 2 hr at room temperature. Target proteins were used with Ez westlumi-plus (ATTO, Tokyo, Japan) and detected by Lumino graph II (ATTO). All protein expression levels were normalized against GAPDH protein level.
Dichloro-dihydro-fluorescein diacetate (DCFH-DA) assay
Intracellular ROS generation in JEG-3 and BeWo cells was processed using a Dichlorodihydrofluorescein (DCFH-DA) assay. JEG-3 and BeWo cells were seeded at 1 × 105 per well in a 6-well plate with 2 mL phenol-free DMEM media containing 10% FBS. After 24 hr, the medium was replaced with new medium containing AN at concentrations of 10-8 M and 10-6 M, after which cells were incubated for an additional 48 hr. During the experiment, H2O2 was used as a positive control to detect ROS formation in JEG-3 and BeWo cells. Specifically, 3% H2O2 solution was added to the well with 1 × 105 cells of JEG-3 and BeWo cells at 1% concentration and then incubated for 30 min. Following incubation, the culture medium was removed and each well was treated with 2 mL DCFH-DA solution in PBS. This plate was subsequently placed in a dark room at room temperature for 30 min, after which the fluorescence intensity of DCF (an oxidized form of DCFH; formed following the uptake and subsequent hydrolysis of DCFH-DA) was measured and photographed using a fluorescence microscope (IX-73 Inverted Microscopy, Olympus, Tokyo, Japan) to detect ROS production. The ROS production in each treatment was quantified using the Cell Sens Dimension software (Olympus).
Assay of terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labeling
The effects of AN on apoptosis of JEG-3 and BeWo cells was measured using a DeadEnd™ fluorometric terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labeling (TUNEL) system (Promega, Madison, WI, USA). The TUNEL assay was processed by detection of the fluorescence of apoptotic cells using TdT enzyme according to the manufacturer’s instructions. Briefly, JEG-3 and BeWo cells were seeded at 1× per well in a 6-well plate with DMEM media. After 24 hr, the medium was changed to new medium with AN at concentrations of 10-8 M and 10-6 M. After 48 hr, the cells were fixed with 4% methanol-free formaldehyde (pH 7.4) for 1 hr, permeabilized using lysis buffer (1% Triton X-100 in 1% sodium citrate) for 15 min, and then treated with 50 μL TdT enzyme buffer, which bound to DNA strand breaks. Finally, labeled strand breaks were identified based on the attachment of fluorescein isothiocyanate-5-dUTP. All wells were counterstained using 4, 6-diamidino-2-phenylindole (DAPI; Invitrogen Life Technologies) and observed under a fluorescence microscope (IX-73 Inverted Microscopy, Olympus), then analyzed using the Image J program. The apoptotic cells produced by each treatment were quantified using the Cell Sens Dimension software (Olympus).
Data analysis
All experiments were conducted at least three times, and all data were analyzed with the Graph-pad Prism software (San Diego, CA, USA). Data were expressed as the means ± SD and analyzed by one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparison test. P-values < 0.05 were considered to be statistically significant.
RESULTS
Acrylonitrile induced choriocarcinoma cell growth arrest
To identify the effects of AN on choriocarcinoma cell viability and appropriate AN concentrations for subsequent experiments, JEG-3 and BeWo cells were cultured with 0.1% DMSO (control) and AN (from 10-9 to 10-3) for 6 days, after which the cell viability was measured using an EZ-cytox kit. The results revealed that AN decreased the cell viability of both cell lines in a dose-dependent manner, and the degree of decline in cell viability by AN appeared larger in BeWo cells than in JEG-3 cells (Fig. 1). According to the cell viability results, two concentrations, 10-8 and 10-6 M of AN, were selected for further experiments.

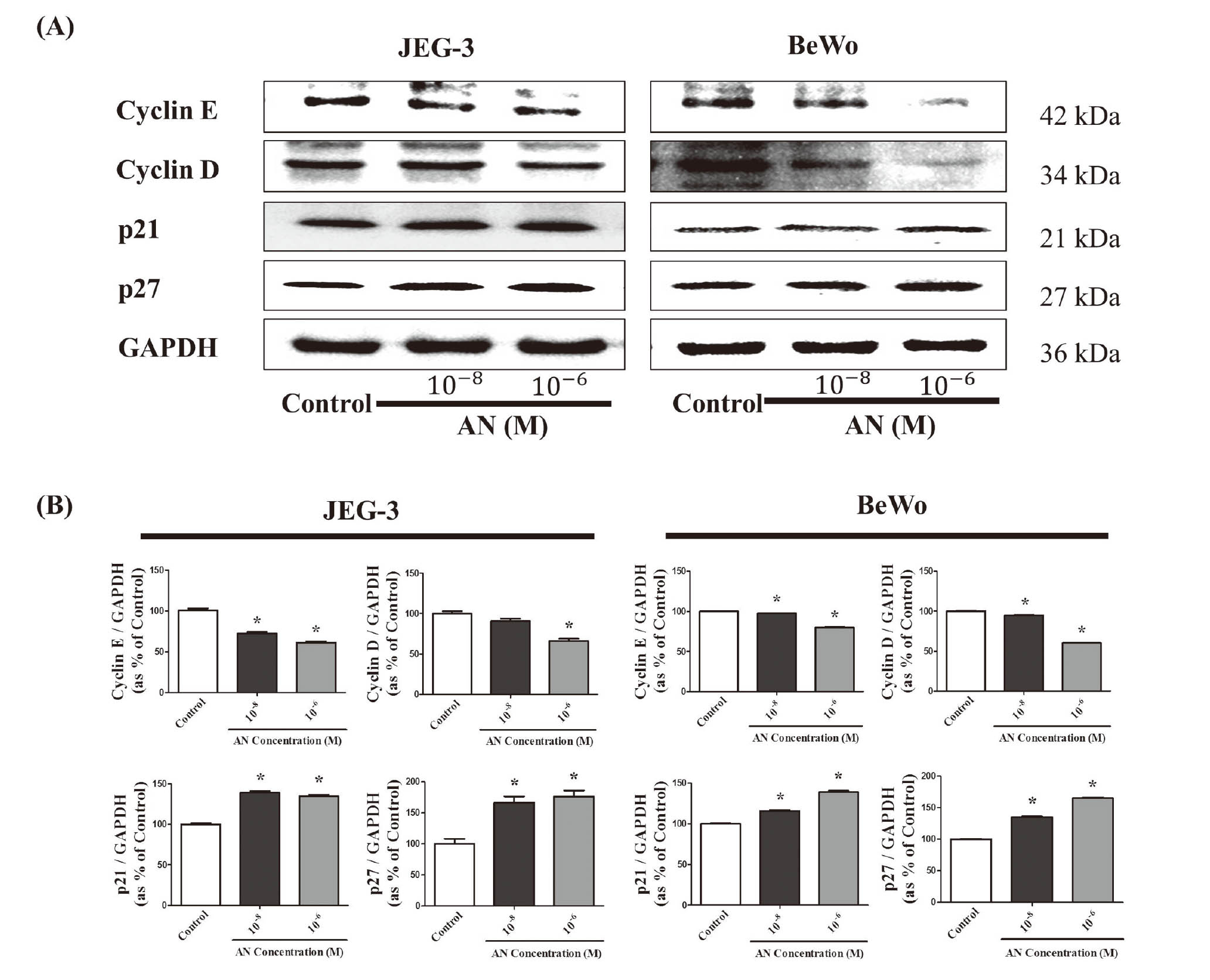
Acrylonitrile was found to decrease expression of cyclin D and cyclin E proteins in a dose-dependent manner, while the expression of p21 and p27 proteins increased in a dose-dependent manner (Fig. 2A). These results indicate that AN suppressed growth of JEG-3 and BeWo choriocarcinoma cells by down-regulating cyclin D and cyclin E and up-regulating p21 and p27 (Fig. 2B).
Acrylonitrile induced apoptosis of choriocarcinoma cells
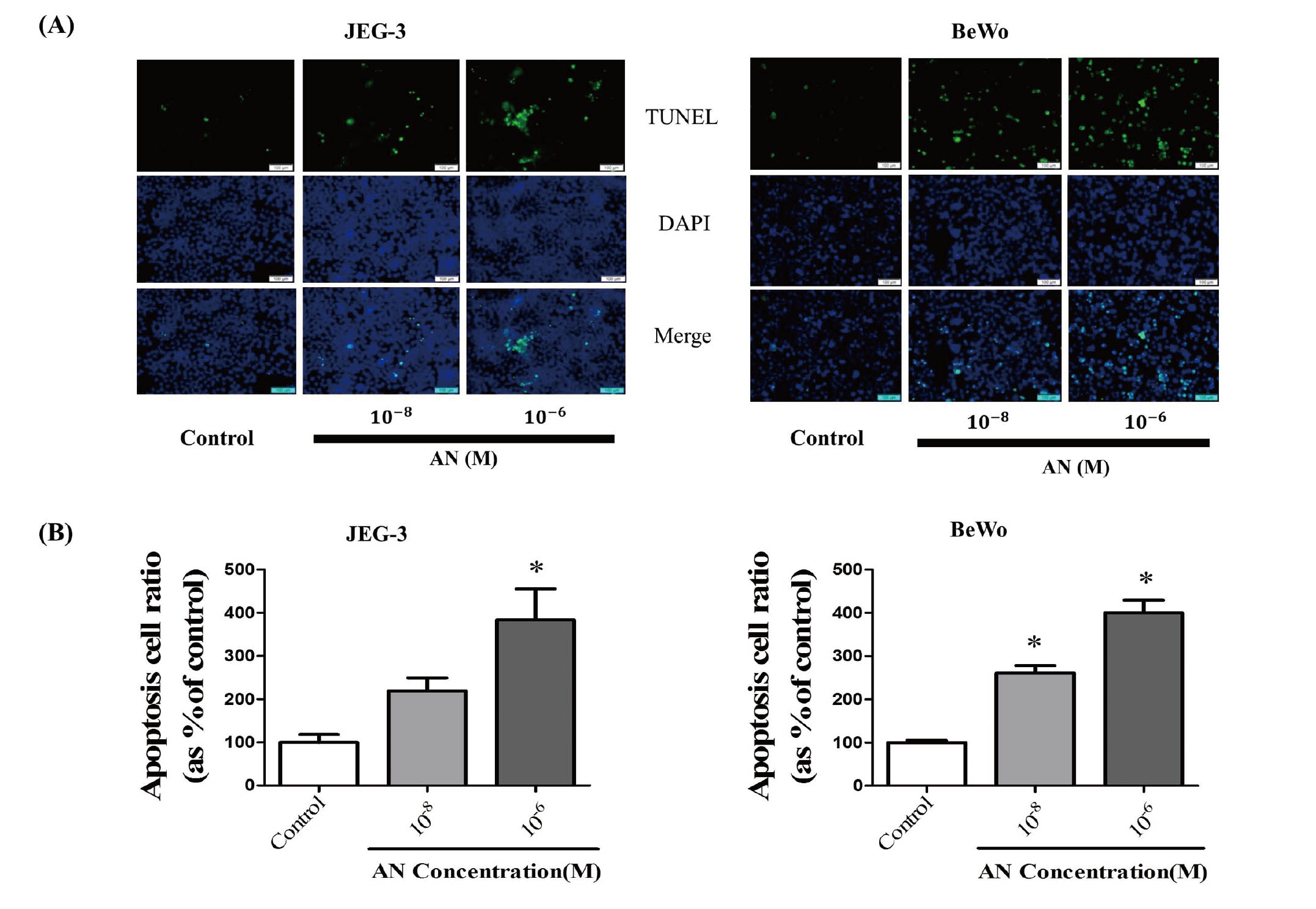
After JEG3 and BeWo cells were treated with AN (10-8 and 10-6M) for 48 hr, AN-induced apoptotic cells were measured by TUNEL assay and then photographed under a fluorescence microscope. The merged images of AN-treated choriocarcinoma cells after the TUNEL reaction and DAPI staining showed that apoptotic cells increased following treatment with AN when compared with the control (Fig. 3A). Integrated optical density analysis also confirmed that the ratio of apoptotic cells in cases of AN exposure was significantly higher than that in the control (Fig. 3B).
Based on the results of the TUNEL assay, the effects of AN on the expression of apoptosis-related genes was investigated by Western blot assay. The p53 protein is a tumor suppressor that is also responsible for senescence or programmed cell death (Kung and Murphy, 2016) and caspase-independent apoptosis (CIA) (Ranjan and Iwakuma, 2016). In this study, p53 was up-regulated by the treatment of AN in a dose-dependent manner (Fig. 4A). The expression of Bax, a pro-apoptotic protein mediating cell death, was increased by treatment with AN in a dose-dependent manner, while Bcl-xl, an anti-apoptotic protein, was decreased by treatment with AN in a dose-dependent manner (Fig. 4A). Caspase 3, a death protease (Porter and Jänicke, 1999), was increased by treatment with AN in a dose-dependent manner (Fig. 4A). These results indicate that AN induced the apoptosis of JEG-3 and BeWo choriocarcinoma cells by up-regulating p53, Bax, and caspase 3 and down-regulating Bcl-xl (Fig. 4B).

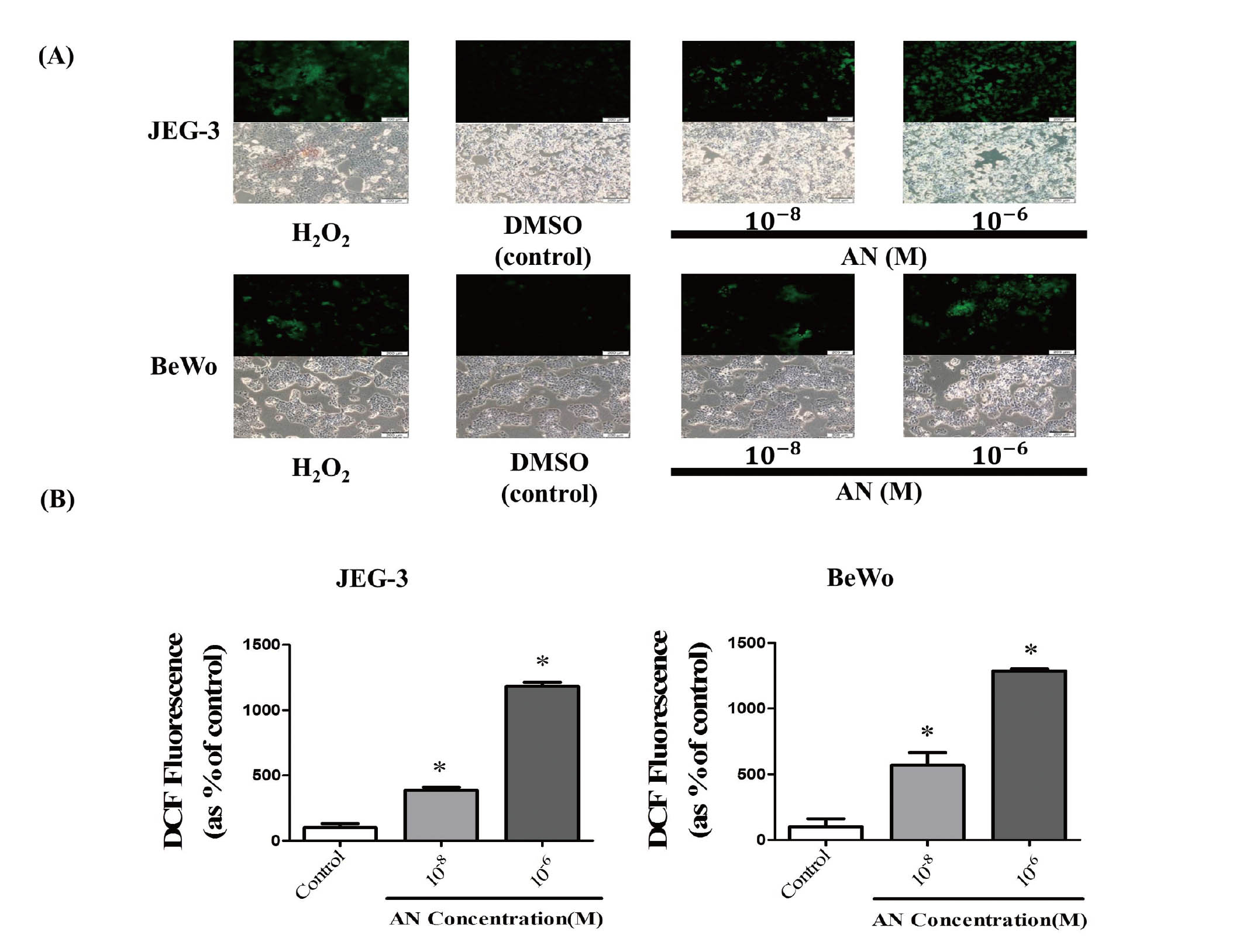
To identify the mechanism of growth arrest and apoptosis occurring in JEG3 and BeWo cells by exposure to AN, we further evaluated the ability of AN to induce oxidative stress via ROS activation. According to previous studies, oxidative stress derived from ROS has been shown to cause cell death in tumor cells through activation of apoptosis (Lennon et al., 1991; Zhao et al., 2016). ROS formation was determined using DCFH-DA. A typical ROS, H2O2, was used as a positive control of ROS formation. Fluorescence microscopy analysis showed that intracellular ROS production in JEG-3 and BeWo cells increased in response to AN exposure (Fig. 5A). The number of DCF positive cells was significantly increased in response to AN exposure for 48 hr (Fig. 5B). These results suggest that increased ROS formation may play an important role in AN-induced oxidative stress and apoptosis of these choriocarcinoma cells.
Acrylonitrile increased protein expression of apoptosis-related factors via ROS formation
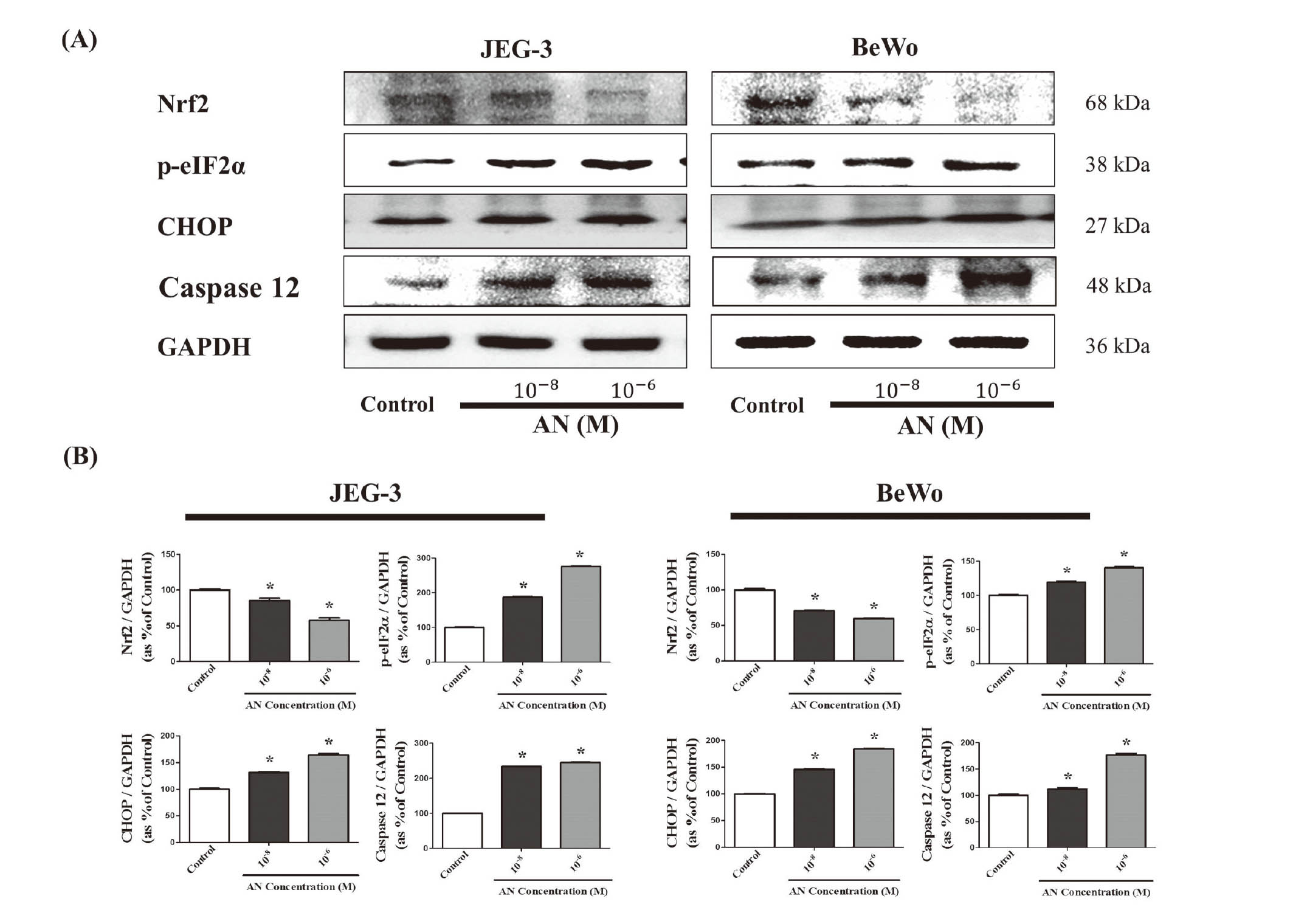
Based on increased formation of ROS by AN, we identified alterations in protein expression of anti-oxidation-related and apoptosis genes associated with ROS activation by Western blot assay. Nuclear factor (erythroid-derived 2)-like 2 (Nrf2), a regulator of cellular resistance to oxidative process (Ma, 2013), was down-regulated by AN treatment for 4 hr in both JEG-3 and BeWo cell lines in a dose-dependent manner (Fig. 6A). Protein expression of phospho-eIF2α (p-eIF2α) and CHOP, a ROS-induced ER-stress marker and an apoptosis gene derived from ER-stress (Li et al., 2014; Wek et al., 2006), was found to be increased by AN exposure in a dose-dependent manner (Fig. 6A). Moreover, protein expression of caspase 12, which is known to be proteolytically activated via ER stress-induced apoptosis (Shiraishi et al., 2006), was increased by AN for 4 hr in a dose-dependent manner (Fig. 6A). These findings indicated that AN caused apoptosis of choriocarcinoma cells via ER stress by ROS activation and down-reduction of an anti-oxidation factor (Fig. 6B).
DISCUSSION
The present study evaluated the potential of cytotoxic effects of AN to induce oxidative stress in JEG3 and BeWo choriocarcinoma cancer cell lines. In the cell viability assay, AN significantly decreased the cell viability of JEG3 and BeWo cells from a quite low concentration, 10-8 M for JEG3 and 10-9 M for BeWo, respectively, in a dose-dependent manner. AN is reported to have a negative impact on health, including headache, insomnia, chest pains, fatigue, general malaise and irritability, in cases of exposure to chronically low concentrations (Canadian Centre for Occupational Health and Safety, 1987). Thus, our study selected the lowest concentrations of AN at which cell survival was significantly reduced to determine the effects at the lowest possible concentration. According to this result of cell viability result, two doses (10-8 and 10-6 M) of AN were selected for further study.
In connection with cytotoxic effect of AN, it was confirmed that AN induced cell cycle arrest by up-regulating the protein expression of p21 and p27 and down-regulating the protein expression of cyclin D and cyclin E (Karimian et al., 2016). The synthesis of cyclin D is initiated during G1 and drives the G1/S phase transition (Choi and Anders, 2014), while cyclin E promotes the expression of related genes that drive cells to the S phase through the G1 phase (Lopez-Beltran et al., 2006). Therefore, cyclin E and cyclin D regulate G1 to S-phase transition of the cell cycle, leading to cellular division. In contrast, p21 and p27 are the regulators of cell cycle progression at the G1 and S phase, which are responsible for the arrest of cell cycle transition (Karimian et al., 2016). As members of the cip/kip family, p21 and p27 can inhibit cell cycle progression by functioning as inhibitors of cyclin D/E kinases (Hawkes et al., 2009), which are required for the G1–S phase transition of the cell cycle (Abukhdeir and Park, 2008; Karimian et al., 2016).
To elucidate the mechanisms by which AN reduces cell viability, we investigated its roles in the induction of apoptosis of JEG-3 and BeWo cells. A TUNEL assay revealed that the number of apoptotic cells was significantly higher in AN exposure groups than the control group. Moreover, AN treatment was confirmed to induce apoptosis by up-regulating the protein expression of pro-apoptotic genes such as p53, Bax, and Caspase-3, as well as by down-regulating the protein expression of the anti-apoptotic gene, Bcl-xl. In response to stimulation of external stress, p53 brings about various cellular responses that can lead to cell-cycle arrest, senescence, differentiation, DNA repair, and apoptosis. Moreover, p53 mediates both membrane permeabilization (MOMP) and permeability transition pore (PTP) opening in response to death stimuli. p53-mediated MOMP is tightly connected to the anti-apoptotic and pro-apoptotic members of the Bcl-2 family proteins. Therefore, cytosolic p53 can translocate to mitochondria, where it can immediately affect anti-apoptotic proteins such as Bcl-2, Bcl-xl and Mcl-1, as well as the pro-apoptotic proteins Bax and Bak (Giono and Manfredi, 2006). Additionally, p53 enhances mitochondrial membrane permeabilization and subsequent release of pro-apoptotic factors from the mitochondria (Dashzeveg and Yoshida, 2015). Taken altogether, these results demonstrate that AN treatment induced cell cycle arrest and apoptosis via increased p53 expression.
In relation to the increase in apoptosis by AN, we also examined its roles in the induction of oxidative stress via ROS formation. Upon DCFH-DA assay, AN treatment engendered ROS creation in JEG-3 and BeWo cells in a concentration-dependent manner, indicating that AN-induced oxidative stress is responsible for cytotoxicity in JEG-3 and BeWo cells. In addition to ROS formation, AN failed to induce antioxidation by decreasing the protein expression of Nrf2 as an antioxidant factor so that excess cellular levels of ROS can induce apoptosis and trigger oxidative stress damage (Halliwell, 2011; Tang et al., 2018). The generation of ROS has been shown to induce modulation of signals mediated by tyrosine phosphorylation/dephosphorylation, sulfoxidation of protamine, and oxidative protein folding in ER. These stressful conditions in ER may trigger disruptions of homeostasis and lead to activation of Unfolded Protein Response (UPR) (Cao and Kaufman, 2014). In some cases, ER stress through formation of ROS has been shown to trigger the UPR (Santos et al., 2009). The UPR is regulated by three ER proteins located in the ER, protein kinase RNA-like endoplasmic reticulum kinase (PERK), activating transcription factor-6 (ATF6), and inositol-requiring protein-1 (IRE1). These proteins bind an ER chaperone, which in turn binds immunoglobulin protein, and remain inactive under stress-free conditions. However, dissociation of these proteins from binding-immunoglobulin protein results in their activation (Bahar et al., 2016). Among the ER located proteins, PERK is correlated with a significant increase in phosphorylation eIF2α (p-eIF2α) and rapid downregulation of global protein synthesis (Liu et al., 2008). This result is linked with attenuation of global protein translation and induction of translation of only selective mRNAs, including activating transcription factor 4 (ATF4) (Blais et al., 2004). Generally, ATF4 is a transcription factor that controls a wide range of genes that play an important role in cell adaptation to stress conditions. Moreover, ATF4 may stimulate the gene encoding CCAAT-enhancer-binding protein homologous protein (CHOP), which is responsible for initiation of the apoptotic cascade (Nishitoh, 2012). The UPR acts to relieve inflammation through the occurrence of ER stress during the cell damage (Tang et al., 2018; Wang et al., 2014). However, if this is not possible, then it sends signals for cell death through apoptosis (Du et al., 2018; Liu et al., 2018). According to results, We also subsequently observed increased p-eIf2α and protein expression of CHOP and caspase 12 in AN-exposed JEG3 and BeWo cells, indicating that AN triggered ER stress-associated apoptosis.
In summary, AN suppressed cell viability of JEG3 and BeWo choriocarcinoma cancer cells by increasing the protein expression of cell cycle arrest genes such as p53 and p21. Moreover, AN induced apoptosis by regulating protein expression of apoptosis-related genes. Increased apoptosis by AN was identified based on its roles in mitochondrial pathway as well as in the induction of ER-stress via ROS production (Fig. 7). Accordingly, AN was confirmed to have a distinct cytotoxicity that occurs by promoting cell cycle arrest and apoptosis via the induction of ROS-mediated ER stress in choriocarcinoma cancer cells. Since JEG-3 and BeWo choriocarcinoma cell lines are derived from the placentas of pregnant women, it can be inferred that AN can significantly induce cytotoxicity of the placenta and disrupt placental function. Furthermore, CS as a main exposure route of AN can also affect placental function. Based on these results, the in vivo effects of AN on the role of the placenta in maintaining a normal state of pregnancy should be investigated in further studies.

ACKNOWLEDGMENTS
This research was supported by grants from the Ministry of Food and Drug Safety in 2020 (19183MFDS463). In addition, this work was also supported by the Global Research and Development Center (GRDC) Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (2017K1A4A3014959).
Conflict of interest
The authors declare that there is no conflict of interest.
REFERENCES
- Abukhdeir, A.M. and Park, B.H. (2008): P21 and p27: roles in carcinogenesis and drug resistance. Expert Rev. Mol. Med., 10, e19.
- Bahar, E., Kim, H. and Yoon, H. (2016): ER Stress-Mediated Signaling: Action Potential and Ca(2+) as Key Players. Int. J. Mol. Sci., 17, 1558.
- Blais, J.D., Filipenko, V., Bi, M., Harding, H.P., Ron, D., Koumenis, C., Wouters, B.G. and Bell, J.C. (2004): Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol. Cell. Biol., 24, 7469-7482.
- Canadian Centre for Occupational Health and Safety. (1987): CCOHS--a national resource for information on occupational health and safety. Dimens. Health Serv., 64, 43-44.
- Cao, S.S. and Kaufman, R.J. (2014): Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal., 21, 396-413.
- Carmody, M., Waszczak, C., Idänheimo, N., Saarinen, T. and Kangasjärvi, J. (2016): ROS signalling in a destabilised world: A molecular understanding of climate change. J. Plant Physiol., 203, 69-83.
- Choi, Y.J. and Anders, L. (2014): Signaling through cyclin D-dependent kinases. Oncogene, 33, 1890-1903.
- Dashzeveg, N. and Yoshida, K. (2015): Cell death decision by p53 via control of the mitochondrial membrane. Cancer Lett., 367, 108-112.
- Du, K., Liu, M., Zhong, X., Yao, W., Xiao, Q., Wen, Q., Yang, B. and Wei, M. (2018): Epigallocatechin Gallate Reduces Amyloid β-Induced Neurotoxicity via Inhibiting Endoplasmic Reticulum Stress-Mediated Apoptosis. Mol. Nutr. Food Res., 62, e1700890.
- Duan, W., Li, S., Meng, X., Sun, Y. and Jia, C. (2017): Smoking and survival of breast cancer patients: A meta-analysis of cohort studies. Breast, 33, 117-124.
- Enongene, E.N., Sun, P.N. and Mehta, C.S. (2000): Sodium thiosulfate protects against acrylonitrile-induced elevation of glial fibrillary acidic protein levels by replenishing glutathione. Environ. Toxicol. Pharmacol., 8, 153-161.
- Esmat, A., El-Demerdash, E., El-Mesallamy, H. and Abdel-Naim, A.B. (2007): Toxicity and oxidative stress of acrylonitrile in rat primary glial cells: preventive effects of N-acetylcysteine. Toxicol. Lett., 171, 111-118.
- Giono, L.E. and Manfredi, J.J. (2006): The p53 tumor suppressor participates in multiple cell cycle checkpoints. J. Cell. Physiol., 209, 13-20.
- Haber, L.T. and Patterson, J. (2005): Report of an independent peer review of an acrylonitrile risk assessment. Hum. Exp. Toxicol., 24, 487-527.
- Halliwell, B. (2011): Free radicals and antioxidants - quo vadis? Trends Pharmacol. Sci., 32, 125-130.
- Hawkes, W.C., Wang, T.T., Alkan, Z., Richter, B.D. and Dawson, K. (2009): Selenoprotein W modulates control of cell cycle entry. Biol. Trace Elem. Res., 131, 229-244.
- Hebestreit, H., Dibbert, B., Balatti, I., Braun, D., Schapowal, A., Blaser, K. and Simon, H.U. (1998): Disruption of fas receptor signaling by nitric oxide in eosinophils. J. Exp. Med., 187, 415-425.
- IARC. (1999): Acrylonitrile. IARC Monogr. Eval. Carcinog. Risks Hum., 71, 43-108.
- Jeon, S.Y., Go, R.E., Heo, J.R., Kim, C.W., Hwang, K.A. and Choi, K.C. (2016): Effects of cigarette smoke extracts on the progression and metastasis of human ovarian cancer cells via regulating epithelial-mesenchymal transition. Reprod. Toxicol., 65, 1-10.
- Kamendulis, L.M., Jiang, J., Zhang, H., deFeijter-Rupp, H., Trosko, J.E. and Klaunig, J.E. (1999): The effect of acrylonitrile on gap junctional intercellular communication in rat astrocytes. Cell Biol. Toxicol., 15, 173-183.
- Karimian, A., Ahmadi, Y. and Yousefi, B. (2016): Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair (Amst.), 42, 63-71.
- Khan, H.A., Alhomida, A.S. and Arif, I.A. (2009): Neurovestibular toxicities of acrylonitrile and iminodipropionitrile in rats: a comparative evaluation of putative mechanisms and target sites. Toxicol. Sci., 109, 124-131.
- Kirman, C.R., Gargas, M.L., Marsh, G.M., Strother, D.E., Klaunig, J.E., Collins, J.J. and Deskin, R. (2005): Cancer dose--response assessment for acrylonitrile based upon rodent brain tumor incidence: use of epidemiologic, mechanistic, and pharmacokinetic support for nonlinearity. Regul. Toxicol. Pharmacol., 43, 85-103.
- Kitamura, M. and Hiramatsu, N. (2010): The oxidative stress: endoplasmic reticulum stress axis in cadmium toxicity. Biometals, 23, 941-950.
- Kung, C.P. and Murphy, M.E. (2016): The role of the p53 tumor suppressor in metabolism and diabetes. J. Endocrinol., 231, R61-R75.
- Lennon, S.V., Martin, S.J. and Cotter, T.G. (1991): Dose-dependent induction of apoptosis in human tumour cell lines by widely diverging stimuli. Cell Prolif., 24, 203-214.
- Li, Y., Guo, Y., Tang, J., Jiang, J. and Chen, Z. (2014): New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim. Biophys. Sin. (Shanghai), 46, 629-640.
- Liu, J., Luo, L.F., Wang, D.L., Wang, W.X., Zhu, J.L., Li, Y.C., Chen, N.Z., Huang, H.L. and Zhang, W.C. (2018): Cadmium induces ovarian granulosa cell damage by activating PERK-eIF2α-ATF4 through endoplasmic reticulum stress. Biol. Reprod., 100, 292-299.
- Liu, L., Wise, D.R., Diehl, J.A. and Simon, M.C. (2008): Hypoxic reactive oxygen species regulate the integrated stress response and cell survival. J. Biol. Chem., 283, 31153-31162.
- Lopez-Beltran, A., MacLennan, G.T. and Montironi, R. (2006): Cyclin E as molecular marker in the management of breast cancer: a review. Anal. Quant. Cytol. Histol., 28, 111-114.
- Ma, Q. (2013): Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol., 53, 401-426.
- Nazaroff, W.W. and Singer, B.C. (2004): Inhalation of hazardous air pollutants from environmental tobacco smoke in US residences. J. Expo. Anal. Environ. Epidemiol., 14 (Suppl. 1), S71-S77.
- Nishitoh, H. (2012): CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem., 151, 217-219.
- Porter, A.G. and Jänicke, R.U. (1999): Emerging roles of caspase-3 in apoptosis. Cell Death Differ., 6, 99-104.
- Ranjan, A. and Iwakuma, T. (2016): Non-Canonical Cell Death Induced by p53. Int. J. Mol. Sci., 17, 2068.
- Ray, P.D., Huang, B.W. and Tsuji, Y. (2012): Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal., 24, 981-990.
- Redza-Dutordoir, M. and Averill-Bates, D.A. (2016): Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta, 1863, 2977-2992.
- Santos, C.X., Tanaka, L.Y., Wosniak, J. and Laurindo, F.R. (2009): Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid. Redox Signal., 11, 2409-2427.
- Satcher, D., Thompson, T.G. and Koplan, J.P. (2002): Women and smoking: a report of the Surgeon General. Nicotine Tob. Res., 4, 7-20.
- Shen, Y.H., Wang, X.L. and Wilcken, D.E. (1998): Nitric oxide induces and inhibits apoptosis through different pathways. FEBS Lett., 433, 125-131.
- Shiraishi, H., Okamoto, H., Yoshimura, A. and Yoshida, H. (2006): ER stress-induced apoptosis and caspase-12 activation occurs downstream of mitochondrial apoptosis involving Apaf-1. J. Cell Sci., 119, 3958-3966.
- Simon, H.U., Haj-Yehia, A. and Levi-Schaffer, F. (2000): Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis, 5, 415-418.
- Sobus, S.L. and Warren, G.W. (2014): The biologic effects of cigarette smoke on cancer cells. Cancer, 120, 3617-3626.
- Sponsiello-Wang, Z., Sanders, E. and Weitkunat, R. (2006): Occupational acrylonitrile exposure and lung cancer: a meta-analysis. J. Environ. Sci. Health Part C Environ. Carcinog. Ecotoxicol. Rev., 24, 257-284.
- Tang, J.Y., Ou-Yang, F., Hou, M.F., Huang, H.W., Wang, H.R., Li, K.T., Fayyaz, S., Shu, C.W. and Chang, H.W. (2018): Oxidative stress-modulating drugs have preferential anticancer effects - involving the regulation of apoptosis, DNA damage, endoplasmic reticulum stress, autophagy, metabolism, and migration. Semin. Cancer Biol., 58, 109-117.
- Verbon, E.H., Post, J.A. and Boonstra, J. (2012): The influence of reactive oxygen species on cell cycle progression in mammalian cells. Gene, 511, 1-6.
- Wang, W.A., Groenendyk, J. and Michalak, M. (2014): Endoplasmic reticulum stress associated responses in cancer. Biochim. Biophys. Acta, 1843, 2143-2149.
- Wek, R.C., Jiang, H.Y. and Anthony, T.G. (2006): Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans., 34, 7-11.
- World Health Organization. (2000): Air quality guidelines for Europe. WHO Reg. Publ. Eur. Ser., V-X, 1-273.
- Yu, B., Wenjun, Z., Changsheng, Y., Yuntao, F., Jing, M., Ben, L., Hai, Q., Guangwei, X., Suhua, W., Fang, L., Aschner, M. and Rongzhu, L. (2016): Preconditioning of endoplasmic reticulum stress protects against acrylonitrile-induced cytotoxicity in primary rat astrocytes: the role of autophagy. Neurotoxicology, 55, 112-121.
- Zhang, L., Connor, E.E., Chegini, N. and Shiverick, K.T. (1995): Modulation by benzo[a]pyrene of epidermal growth factor receptors, cell proliferation, and secretion of human chorionic gonadotropin in human placental cell lines. Biochem. Pharmacol., 50, 1171-1180.
- Zhao, M., Zhu, P., Fujino, M., Zhuang, J., Guo, H., Sheikh, I., Zhao, L. and Li, X.K. (2016): Oxidative Stress in Hypoxic-Ischemic Encephalopathy: Molecular Mechanisms and Therapeutic Strategies. Int. J. Mol. Sci., 17, E2078.