Abstract
Bisphenol A (BPA) interferes the function and development of the central nervous system (CNS), resulting in behavioral abnormalities and memory loss. S-nitrosylation of protein disulfide isomerase (PDI) is increased in brains with sporadic Alzheimer’s disease and Parkinson’s disease. The aim of the present study was to clarify the role of nitric oxide (NO) in BPA-induced neurotoxicity. Since rotenone induces NO-mediated neurodegeneration through S-nitrosylation of PDI, it was used as a positive control. First, rats were treated with BPA and rotenone, and S-nitrosylation of PDI was detected in rat brain microsomes. BPA and rotenone decreased RNase oxidation activity of PDI concomitant with S-nitrosylation of PDI. Next, to clarify S-nitrosylation of PDI by BPA and rotenone in rat brains, we treated the rat pheochromocytoma cell line PC12 and primary cultured neuron cells from the rat hippocampus with BPA (5 and 10 μM) and rotenone (100 or 200 nM). BPA induced S-nitrosylation of PDI, while NG-monomethyl-L-arginine (L-NMMA), a NOS inhibitor, exerted the opposite effects. Finally, to evaluate the toxicity of BPA in the CNS, we investigated its effects on neurite outgrowth of PC12 and primary cultured neuron cells. BPA inhibited neurite outgrowth of these cells, while L-NMMA reversed this inhibition. The involvement of PDI activity in neurite outgrowth was also examined, and bacitracin, a PDI inhibitor, is shown to decrease neurite outgrowth. Furthermore, the overexpression of PDI, but not a catalytically inactive PDI mutant, enhanced neurite outgrowth. These results suggested that S-nitrosylation of PDI induced by excessive NO caused BPA-induced neurotoxicity.
INTRODUCTION
Bisphenol A (BPA) is an environmental pollutant that disrupts the endocrine system (Kim et al., 2009; Fujimoto and Aou, 2018). BPA binds to estrogen receptor (ER) and acts as an agonist (Bruno et al., 2019). It has also been shown to disrupt the central nervous system (CNS). In the early development of Xenopus laevis, BPA induces apoptosis of cells in the CNS (Oka et al., 2003). Mice prenatally exposed to BPA exhibit functional disorders in the CNS and hyperactivity (Suzuki et al., 2003). Furthermore, BPA decreases the ratio of GABAergic neurons in cortical neurospheres from embryonic mice (Fukushima and Nagao, 2018). Maternal exposure to BPA is found to alter the white matter microstructure, leading to internalizing behavior in children (Grohs et al., 2019). BPA is also shown to decrease spatial memory and dendritic spine density in ovariectomized female rats (Bowman et al., 2019). Although recent studies have implicated BPA exposure in neurotoxicity, the underlying molecular mechanisms have not yet been elucidated.
We previously reported that BPA increases nitric oxide (NO) levels in the human hepatoma cell line Hep3B, while NG-monomethyl-L-arginine (L-NMMA) reduces these BPA-induced increases (Nakamura et al., 2018). Moreover, BPA induces S-nitrosylation of kelch-like ECH-associated protein (Keap1) (Nakamura et al., 2018). Previous studies demonstrated that BPA increases NO levels by activating nitric oxide synthase (NOS) (Chouhan et al., 2015; Noguchi et al., 2002). NO is necessary for the physiological function of the brain, whereas excessive NO has been associated with brain dysfunctions (Nakamura and Lipton, 2011). Excessive NO is one of the important pathways leading to neuronal cell death in neurodegenerative diseases, including Alzheimer’s diseases and Parkinson’s disease (Nomura, 2004; Nakamura et al., 2013). BPA is shown to increase NO levels and affects synaptic activity in the rat spinal cord, resulting in neurobehavioral disorders (Pandey and Deshpande, 2015).
A recent study identifies S-nitrosylation of protein disulfide isomerase (PDI) as an underlying mechanism for NO-induced neurotoxicity (Uehara et al., 2006). PDI is an enzyme for protein folding that catalyzes the oxidation, reduction, and isomerization of the disulfide bonds of misfolded or nascent proteins (Vuori et al., 1992). PDI is also a neuroprotective protein. The silencing of PDI increases apoptosis in neuron cells under endoplasmic reticulum (ER) stress (Honjo et al., 2017). The up-regulation of PDI has been shown to increase the viability of neuroblastoma cells in the ischemic rat brain (Tanaka et al., 2000). A PDI inhibitor decreases the number of neurons and increases the aggregation of orexin-immunoreactive neurons (Obukuro et al., 2013). The up-regulation of NO in BPA-treated rats is found to disrupt the thyroid-brain axis, which inhibits brain development (Ahmed et al., 2018). Thyroid hormone also binds to PDI, which inhibits growth hormone release via thyroid hormone receptor (Okada et al., 2007).
Importantly, PDI is S-nitrosylated in brains with sporadic Alzheimer’s disease and Parkinson’s disease, which are NO-mediated neurodegenerative disorders that are characterized by abnormal protein accumulation. A previous study reported that S-nitrosylation of PDI decreases its chaperone and isomerase activities, and, as a result, leads to PDI dysfunction (Uehara et al., 2006). S-nitrosylated PDI induces the aggregation of synphilin, a component of the Lewy bodies observed in the brains of patients with Parkinson’s disease (Uehara et al., 2006). S-nitrosylation of PDI is shown to promote the accumulation of polyubiquitinated proteins, which induces the unfolded protein response (UPR) and contributes to cell death (Uehara et al., 2006). Another study reported that a NO donor induces S-nitrosylated PDI and decreases its activity in the hypothalamus of mice (Obukuro et al., 2013). Furthermore, decreases in PDI activity induces the aggregation of proteins and degeneration of orexin neurons, and endoplasmic reticulum stress (ER stress) is suggested to play a role in neuron degeneration (Obukuro et al., 2013). Therefore, we hypothesized that S-nitrosylation of PDI is involved in BPA-induced neurotoxicity.
Rotenone is neurotoxic compound as well as BPA. Rotenone is a mitochondrial complex I inhibitor that induces neurodegeneration and Parkinson’s disease-like symptoms (Emmrich et al., 2013; Betarbet et al., 2000; Li et al., 2003). Rotenone increases NO levels, and excessive NO is involved in rotenone-induced neurodegeneration (Xiong et al., 2015; He et al., 2003). NO induced by rotenone induces S-nitrosylation of PDI in SH-SY5Y cells, leading to neural toxicity (Uehara et al., 2006). Therefore, to investigate the involvement of S-nitrosylated PDI in neuron degeneration induced by BPA, rotenone was used as a positive control.
In the present study, to elucidate the mechanisms underlying the neurotoxicity induced by BPA, we used PC12 cells. PC12 cells are a model system for neurodegenerative diseases and a well-established model for neurite outgrowth induced by NGF (Greene and Tischler, 1976). Neurite outgrowth is important in nervous system development (Hjorth et al., 2014). The differentiation of neurite outgrowth induces the extension and branching of axons, which are required for connections with their targets, resulting in the establishment of neuronal networks (Hjorth et al., 2014; Zschätzsch et al., 2014). Neurite outgrowth is suppressed by a number of compounds, such as rotenone and colchicine, which interrupt the function of the CNS, and is enhanced by inhibitors of the Rho kinase pathway (Krug et al., 2013), (Negishi and Katoh, 2002). Moreover, neurite outgrowth is inhibited by CNS myelin, which reduces axonal regeneration (Ng et al., 1996). Therefore, we examined neurite outgrowth to evaluate the toxic effects of BPA on the function of the CNS. We also investigated the role of S-nitrosylation of PDI in BPA-induced neurotoxicity.
MATERIALS AND METHODS
Materials
Fetal bovine serum, penicillin-streptomycin solution, S-methyl methanethiosulfonate (MMTS), and rotenone were purchased from Sigma (St. Louis, MO, USA). Horse serum was from Equitech-Bio Inc. (Kerrville, TX, USA). (N-[6-(biotinamido)hexyl]-3’-(2’-pyridyldithio)propionamide (Biotin-HPDP) and streptavidin-agarose were purchased from Pierce Biotechnology (Rockford, IL, USA). Nerve growth factor (NGF) was purchased from Alomone Labs (Jerusalem, Israel). Dulbecco’s modified Eagle’s medium (DMEM), bisphenol A, bisphenol F, bacitracin, NOC7, and L-NMMA were obtained from Wako Pure Chemical Industries, Ltd. (Osaka, Japan).
Animal treatment and preparation of rat brain microsomes
Male Sprague-Dawley rats at postnatal day 7 weeks were purchased from SLC Inc. (Shizuoka, Japan). Rats were injected intraperitoneally with BPA at 100 mg/kg/day or rotenone at 3 mg/kg/day (Luo et al., 2007) dissolved in corn oil daily for 6 days. To prepare rat brain microsomes, rat brains were homogenized with 0.25 M sucrose solution, and then centrifuged at 8000 × g for 10 min followed by ultracentrifugation at 100,000 × g for 60 min. The resulting residue was obtained as microsomes. PDI protein levels were measured in microsomes by Western blotting with standards of each purified protein (Miyake et al., 2014). All experiments were conducted in accordance with guidelines on the welfare of experimental animals and with the approval of the Ethics Committee on the use of animals of Kwansei Gakuin University.
Cell culture and cell neurite extension
A rat pheochromocytoma cell line (PC12) was obtained from the RIKEN Cell Bank (Ibaraki, Japan). Cells were maintained in DMEM containing 10% horse serum, 5% fetal bovine serum, penicillin (100 units/mL), and streptomycin (100 μg/mL). Cells were incubated at 37°C in a humidified atmosphere of 5% CO2/95% air. In measurements of neurite outgrowth, PC12 cells were seeded at 1 × 104 per well on 24-well plates. After one day, culture medium was changed to DMEM containing NGF (100 ng/mL) with or without BPA. After 24 hr, neurite length was measured with ImageJ as previously described (Oguro et al., 2018). To overexpress Venus-PDI WT or a cysteine mutant, in which cysteine (55, 58, 399, and 402) was substituted by alanine, WT or mutant PDI cDNA in the pcDNA3.1 vector was lipofected into cells with the transfection reagent Screen Fect A plus (Wako). Images were obtained by confocal microscopy, Nikon A1 (Nikon, Tokyo, Japan), and the neurite length of Venus-expressing cells was measured. Differentiated cells with neurites were defined as those with a neurite length greater than the cell body of an individual cell.
Isolation of rat hippocampal neuronal cells
Neuron-rich cultures were prepared from the hippocampus of Wistar rats (Clea Japan, Inc., Tokyo, Japan) on embryonic day 18 as described previously (Banker and Cowan, 1977). Cells were plated at 1 × 105 cells per cloning ring on polyethyleneimine-coated dishes with Neurobasal Medium (Thermo Fisher Scientific, Waltham, MA, USA) containing 2% B27 Supplement (Thermo), penicillin (100 units/mL) (Thermo), streptomycin (100 μg/mL) (Thermo), 5 μg/mL insulin (Sigma), and 0.5 mmol L−1 L‐glutamine. After 24 hr, the cloning ring was removed, and BPA (5 or 10 μM) was added to the cell medium. After 24 hr, the lengths of neurites that diffused into the free space from cells were measured. All experiments were conducted in accordance with the guidelines on the welfare of experimental animals and with the approval of the Ethics Committee on the use of animals of Kwansei Gakuin University.
Biotin-switch assay
Rat brain microsomes, PC12 cells, or rat primary cultured neurite cells were solubilized in HENC buffer (HEN buffer containing 0.4% CHAPS) (HEN buffer: 250 mM Hepes, pH 7.5, 1 mM EDTA, and 0.1 mM neocuproine) on ice for 30 min, and centrifuged at 13,000 × g at 4°C for 5 min. A one-tenth volume of 25% SDS and 20 mM MMTS were added to the supernatant or purified PDI proteins and incubated at 50°C for 20 min with frequent vortexing to block free thiols. The purification of PDI proteins expressed in E. coli was as previously described (Miyake et al., 2014). To remove excess MMTS, proteins were precipitated with 2 volumes of pre-chilled (-20°C) acetone, incubated at -20°C for 2 hr, and centrifuged at 13,000 × g for 5 min, followed by gentle rinsing of the pellet 3 times with 70% acetone. The precipitate was dissolved in 0.2 mL of HENS buffer (HEN buffer containing 1% SDS), 2 μL of 100 mM ascorbate and 20 μL of 10 mM biotin-HPDP (prepared freshly in N,N-dimethylformamide) were added, and cells were incubated at room temperature for 1 hr. Until this point, all procedures were performed in the dark. Samples were precipitated with acetone and resuspended in 0.2 mL of HENS buffer. Samples ware diluted in 2 volumes of neutralization buffer (20 mM HEPES, pH 7.5, 0.1 M NaCl, 1 mM EDTA, and 0.5% Triton X-100). Biotinylated proteins were incubated with 20 μL of 50% (v/v) streptavidin-agarose beads (Pierce) at room temperature for 1 hr or at 4°C overnight with continuous gentle inversion agitation. Samples were then centrifuged at 6,000 × g for 1 min, and the supernatant containing unbound proteins was discarded. Beads were washed 5 times with washing buffer (20 mM HEPES, pH 7.5, 600 mM NaCl, 1 mM EDTA, and 0.5% Triton X-100). To elute bound proteins, the beads were incubated with 10 μL of elution buffer (20 mM HEPES, pH 7.7, 0.1 M NaCl, 1 mM EDTA, and 100 mM 2-mercaptoethanol) at 37°C for 20 min with gentle agitation. Supernatants were collected by centrifugation, separated on SDS-PAGE, and subjected to a Western blot analysis.
RNase oxidation activity
RNase oxidation activity was assayed as described previously (Hashimoto et al., 2008). Briefly, reduced and denatured RNase A (8 μM) was incubated with rat brain microsomes (150 μg) or purified PDI proteins (100 μg) in 100 mM sodium phosphate buffer, pH 7.5, containing 4.5 mM cytidine 2’, 3’-cyclic monophosphate (cCMP), 2 mM EDTA, 1 mM GSH, and 0.2 mM GSSG at room temperature. The reaction was started by adding reduced and denatured RNase A. Changes in absorbance at 296 nm were monitored with the EnVision 2104 Multilabel Reader spectrophotometer (Perkin Elmer, Foster City, CA, USA).
Statistical analysis
Values are given as the means ± S.D.. Statistical analyses were performed using the Student’s t-test, and p < 0.05 and p < 0.01 were considered to be significant.
RESULTS
S-nitrosylation of PDI in rat brain microsomes or rat primary cultured neurons treated with BPA and rotenone
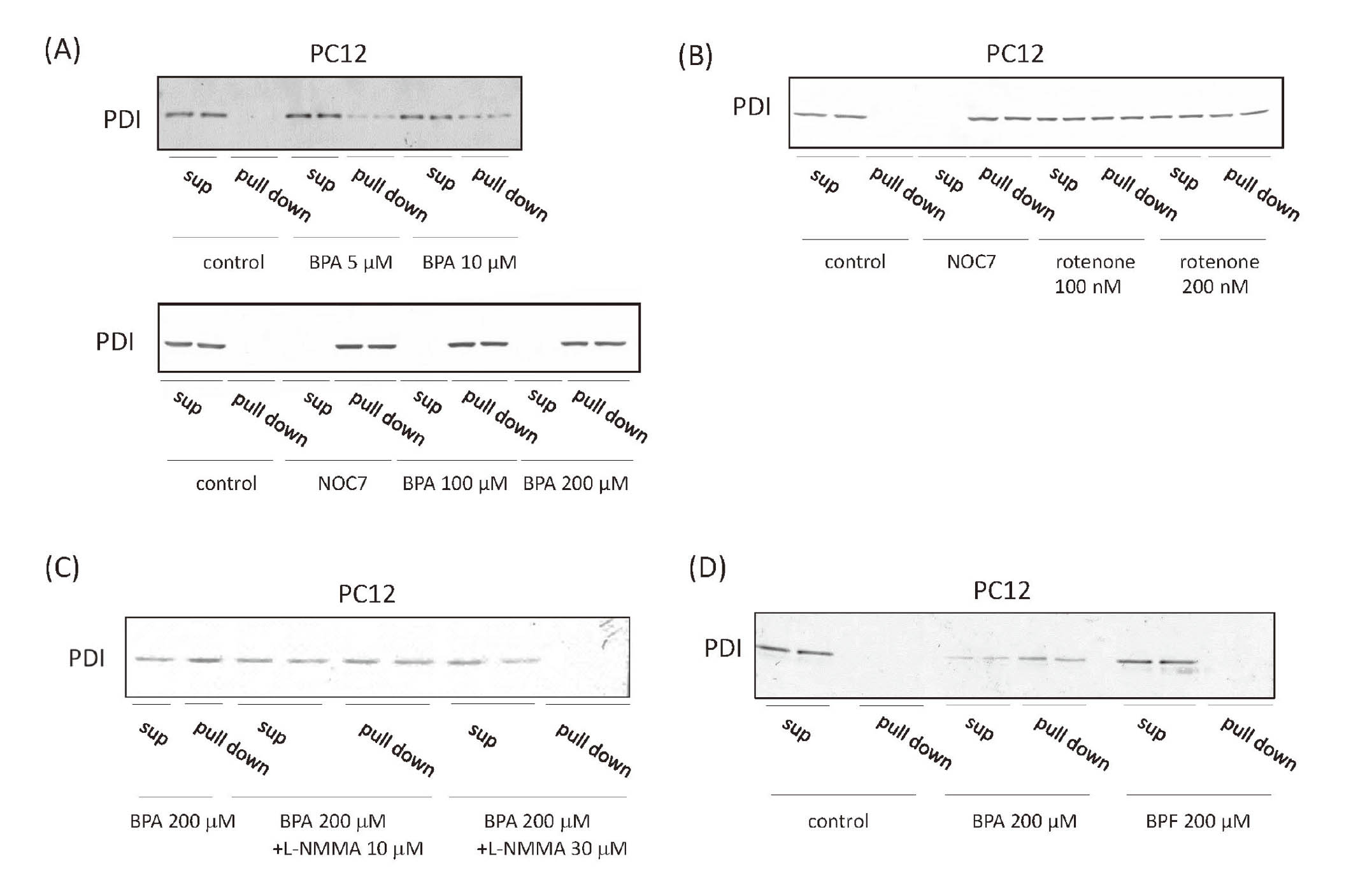
First, we investigated the effects of BPA or rotenone on S-nitrosylation of PDI in rat brain. In the present study, rats were injected intraperitoneally with BPA at 100 mg/kg/day. Another study suggested that 100 mg/kg/day BPA shows toxicity such as apoptosis and sub-G1 cell population (Bilgi et al., 2019; Urriola-Muñoz et al., 2014). On the other hand, rotenone is well known as mitochondrial complex I inhibitor and induces neurodegeneration and Parkinson’s disease-like symptoms (Emmrich et al., 2013). Rotenone induces S-nitrosylated PDI in SH-SY5Y cells, indicating that it causes neurodegeneration via S-nitrosylation of PDI (Uehara et al., 2006). To confirm the induction of S-nitrosylation, we used rotenone as positive control. We found that BPA and rotenone induced S-nitrosylation of PDI in the rat brain microsomes (Fig. 1A). RNase oxidase activity was used as a tool for evaluating PDI oxidative activity. RNase was denatured by pre-treatment with dithiothreitol and guanidine. PDI restored conformation of RNase, and RNase could oxidize cCMP and product CMP. Therefore, we could evaluate PDI oxidative activity by investigating RNase oxidative activity. RNase oxidation activity of rat brain microsomes was decreased by the BPA and rotenone treatment (Fig. 1B). To confirm direct S-nitrosylation of PDI and consequent inhibition of its RNase oxidation activity, purified PDI was treated with the NO donor, NOC7. S-nitrosylated PDI protein levels were increased by the treatment with NOC7 (Fig. 1C), and RNase oxidation activity was decreased (Fig. 1D), suggesting that NOC7 induced S-nitrosylation of PDI and inhibited PDI activity. We then treated primary cultured neurons from the rat hippocampus with rotenone to confirm whether it directly induces S-nitrosylation of PDI. BPA induced S-nitrosylation of PDI in these cells (Fig. 1E). Rotenone also induced S-nitrosylation of PDI in these cells (Fig. 1F), suggesting that BPA activated NOS in neural cells.

We investigated the effects of BPA on S-nitrosylation of PDI in PC12 cells (Fig. 2A). 5 or 10 μM BPA induced S-nitrosylation of PDI, and BPA induced it even in a high concentration (100 or 200 μM). For further investigation of S-nitrosylation of PDI by BPA treatment, PC12 cells were treated with rotenone that induces S-nitrosylation of PDI (Uehara et al., 2006). S-nitrosylation of PDI was induced in PC12 cells by 100 or 200 nM rotenone treatment (Fig. 2B). 30 μM L-NMMA, a NOS inhibitor, efficiently inhibited the S-nitrosylation of PDI, even at high BPA concentrations (100 or 200 μM), in PC12 cells (Fig. 2C), suggesting that BPA activated NOS and induced S-nitrosylated PDI. BPF lacks two methyl groups between the phenol groups in BPA and is used in the manufacture of plastics as a substitute (Yin et al., 2019). However, the effects of BPF on neural differentiation remain unclear. We compared the effects of BPF and BPA on S-nitrosylation of PDI and found that 200 μM BPF did not exert any significant effects (Fig. 2D), indicating that these methyl groups (or hydrophobicity) were important in formation of S-nitrosylated PDI.
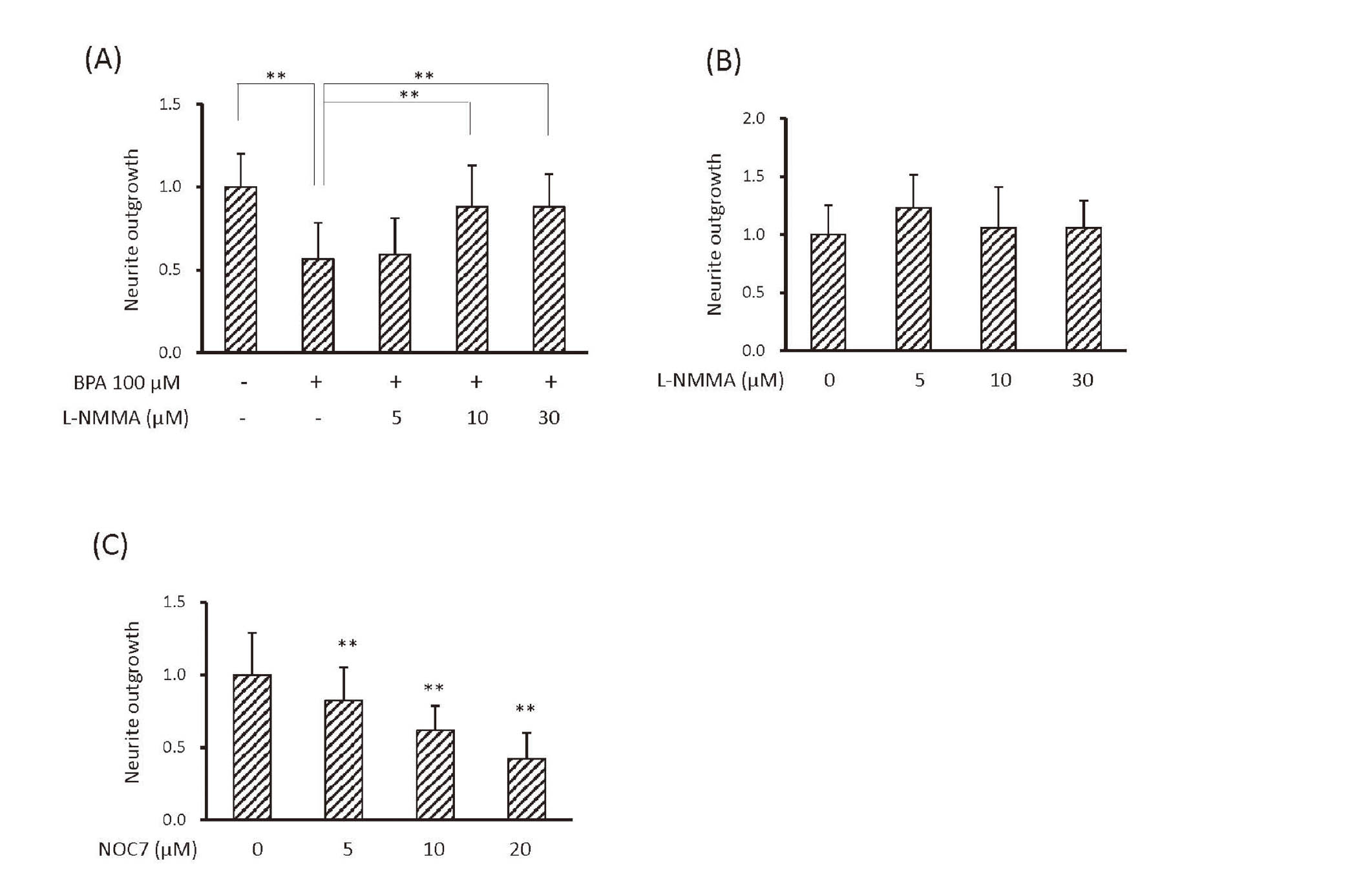
Investigations on neurite outgrowth are important for defining the effects of chemicals on neural function (Krug et al., 2013). BPA (5 or 10 μM) significantly induced S-nitrosylation of PDI and inhibited neurite outgrowth of PC12 cells (Fig. 3A). These results were consistent with previous findings showing that BPA (10 μM) inhibited neurite outgrowth (Wang et al., 2019; Wu et al., 2016). BPA could change neurite outgrowth and S-nitrosylation of PDI at low concentrations (5 or 10 μM). We also investigated the effects of BPA on neurite outgrowth of rat primary cultured neurons at the same concentration, and the results obtained showed that it inhibited neurite outgrowth of rat primary cultured neurons as well as PC12 cells (Fig. 3B). Since 100 or 200 nM rotenone induced the S-nitrosylation of PDI, the inhibition of NGF-induced neurite outgrowth of PC12 cells by 100-300 nM rotenone was investigated. Even 100 nM rotenone significantly inhibited neurite outgrowth of PC12 cells (Fig. 3C). Both BPA and rotenone inhibited neurite outgrowth of PC12 cells, indicating that BPA and rotenone induced S-nitrosylation of PDI and inhibited neurite outgrowth. These results suggested the potential of PC12 cells as a tool for investigating NO-mediated neurotoxicity through S-nitrosylation of PDI.

NO plays an important role in the suppression of neurite outgrowth by BPA (Wang et al., 2019). We also found that BPA-induced S-nitrosylation of PDI was reduced by L-NMMA. BPA (100 μM) inhibited neurite outgrowth of PC12 cells, similar to BPA (5 or 10 μM), and this inhibition was recovered by L-NMMA (Fig. 4A). L-NMMA did not affect basal neurite outgrowth of PC12 cells (Fig. 4B), suggesting that NO synthesis was involved in the effects of BPA on neurite outgrowth. We also examined the effects of NOC7 on neurite outgrowth and found that NOC7, similar to BPA, inhibited neurite outgrowth in a dose-dependent manner (Fig. 4C).
Effects of PDI activity on NGF-induced neurite outgrowth of PC12 cells
S-nitrosylation of PDI suppressed its function. To confirm the contribution of PDI dysfunction to neurite outgrowth of PC12 cells, bacitracin, an inhibitor of PDI, was used. Bacitracin inhibited NGF-induced neurite outgrowth of PC12 cells in a dose-dependent manner (Fig. 5A). To investigate the effects of thiol groups in active sites of PDI on neurite outgrowth, we prepared a fusion protein of Venus with wild type (WT) or a catalytically inactive PDI mutant, in which cysteine residues were substituted by alanine residues at active sites, and overexpressed them in PC12 cells. Overexpressed PDI proteins were easily detected by fluorescence, and we compared neurite outgrowth of fluorescent cells and cells overexpressing Venus only as a control. The overexpression of wild PDI enhanced, while mutant PDI decreased neurite outgrowth (Fig. 5B). These results indicated that the isomerase activity of PDI was required for neurite outgrowth of PC12 cells.
DISCUSSION
In the present study, we found that BPA induced S-nitrosylation of PDI. We detected S-nitrosylation of PDI in brain microsomes of rats treated with BPA, PC12, and rat primary cultured neuronal cells. S-nitrosylation of PDI by BPA were not observed in the presence of L-NMMA, a NOS inhibitor. Furthermore, we found that BPA inhibited neurite outgrowth by inhibiting PDI activity. PDI oxidative activity was inhibited in brain microsomes of rats treated with BPA. We found that BPA inhibited neurite outgrowth of PC12 cells and rat primary cultured neuronal cells, and the inhibition was not observed in the presence of L-NMMA.
BPA induces embryonic and developmental toxicity. It is considered to affect important signaling pathways via ER or kinase signaling receptors, leading to abnormalities in neural development (Ge et al., 2014; Baba et al., 2009). BPA has been shown to attenuate neurite extension (Seki et al., 2011; Liang et al., 2020), branching (Wu et al., 2016), neuron quantities (Zhang et al., 2020), and differentiation (Kim et al., 2009; Tiwari et al., 2015). We also found that BPA inhibited neurite outgrowth of PC12 cells and rat primary cultured neuronal cells. These results were consistent with previous findings showing that BPA inhibited neurite outgrowth (Wang et al., 2019; Wu et al., 2016; Seki et al., 2011). However, limited information is currently available on the mechanisms underlying neuronal abnormalities induced by BPA.
PDI is an enzyme to help protein folding by catalyzing the oxidation, reduction, and isomerization of the disulfide bonds of misfolded or nascent proteins. UPR is necessary for the normal development of cortical neurons and cerebellar white matter (Naughton et al., 2019). It protects neuron cells against ER stress induced by the accumulation of misfolded proteins (Naughton et al., 2019). PDI is one of the critical factors in UPR, and its dysfunction causes the accumulation of unfolded/misfolded proteins in neuronal cells and neurodegeneration. The S-nitrosylation of PDI is associated with synphilin misfolding in Parkinson’s disease, the accumulation of polyubiquitinated proteins (Uehara et al., 2006), and alpha-synuclein misfolding (Wu et al., 2014). Since synphilin and alpha-synuclein are both important in neurite outgrowth (Liu et al., 2013; Li et al., 2010), S-nitrosylation of PDI induced by BPA may inhibit neurite outgrowth via the misfolding of synphilin and alpha-synuclein. In the present study, NO induced S-nitrosylation of PDI and inhibited PDI oxidation activity in vitro. The catalytically inactive PDI mutant inhibited neurite outgrowth of PC12 cells. These results indicated that excess NO induced by BPA inhibited PDI isomerase activity via S-nitrosylation, thereby suppressing neurite outgrowth.
BPA (50-150 μM) has been shown to enhance dopamine release via N-type Ca2+ channels in PC12 cells (Yoneda et al., 2003). Previous studies reveal the neural toxicity in a µM BPA concentration. BPA (100 μM) inhibits basal L-type channel currents and the proliferation of hippocampal neural stem cells (Feiteiro et al., 2018; Agarwal et al., 2016). BPA (less than 200 μM) does not induce cell death, while 200 μM BPA inhibits neurite outgrowth (Cho et al., 2018). In the present study, we used lower concentrations compared with these studies and found that even 5 μM BPA induced S-nitrosylation of PDI in PC12 cells and rat primary cultured neuron cells, leading to the inhibition of neurite outgrowth of PC12 cells.
BPA activates ER alpha, a nuclear ER, and G protein-coupled receptor 30 (GPR30), a membrane ER. The half-maximal inhibitory concentration (IC50) of BPA to ER alpha is shown to be 1030 nM, whereas that of 17 beta-estradiol is 0.88 nM (Matsushima et al., 2010). These findings suggest that 5 μM BPA was relevant concentration that we used in the present study. A recent study reported that 1 nM BPA increases Ca2+ influx via GPR30 (Gonçalves et al., 2018). The binding affinity of BPA to ER alpha is approximately 1,000-fold lower than that to GRP30. These findings indicate that BPA may be concentrated in the transmembrane. In the present study, we examined the effects of BPF, which is less hydrophobic than BPA, on S-nitrosylation of PDI and neurite outgrowth of PC12 cells. BPA derivatives, such as BPF, are replacing BPA because of its toxicity; however, the effects of BPF on neuronal differentiation are still being debated (Yin et al., 2019; Liang et al., 2020; Fujiwara et al., 2018; Rosenfeld, 2017). BPF and BPA cross the blood-brain barrier and accumulate in brain tissue (Charisiadis et al., 2018). BPF also binds to ER (Molina-Molina et al., 2013). However, we found that BPF did not affect S-nitrosylation of PDI or neurite outgrowth (data not shown) from PC12 cells. A previous study demonstrated that BPAF is a more hydrophobic derivative than BPA and has a lower IC50 to ER alpha (53.4 nM) (Matsushima et al., 2010). These findings suggest that BPA may be concentrated in the transmembrane due to its hydrophobicity.
Since rotenone induces neurodegeneration via S-nitrosylation of PDI, we used rotenone as a positive control (Uehara et al., 2006). Rotenone inhibits electron flux between mitochondrial complex I and ubiquinone and the oxidation of NADH, leading to the decrease of ATP production (Xiong et al., 2012). This inhibition also induces reactive oxygen species (ROS) (Xiong et al., 2012). Another study demonstrated that rotenone decreases dopamine transporter associated with dopamine release, resulting in increases in intracellular dopamine and ROS levels in PC12 cells (Sai et al., 2008). ROS activates Ca2+ channels (Görlach et al., 2015). Uehara et al. (2006) showed that rotenone induces S-nitrosylation of PDI in human neuroblastoma cell line (SH-SY5Y cells). We also found that rotenone induced S-nitrosylation of PDI in PC12 cells and rat primary cultured neuron, and S-nitrosylation of PDI attenuated RNase oxidation activity. These results were consistent with previous findings of Uehara et al. (2006). Additionally, rotenone inhibited neurite outgrowth of PC12 cells. These results suggested that PC12 cells were an available tool for the investigation of NO-mediated neurotoxicity through S-nitrosylation of PDI, and S-nitrosylation of PDI also occurred in normal neural cells.
We previously demonstrated that BPA increases NO levels in Hep3B cells (Nakamura et al., 2018). In the present study, BPA did not induce S-nitrosylation of PDI in the presence of L-NMMA, which inhibits all three types of NOS, namely, nNOS, eNOS, and iNOS. PC12 cells express eNOS and nNOS. These two NOS types are activated by Ca2+ via calmodulin. We also found that the influx of Ca2+ increased in PC12 cells treated with BPA (data not shown), suggesting that BPA activated NOS. BPA has been shown to bind to various Ca2+ channels and induce the influx of Ca2+. Transient receptor potential cation channel subfamily V member 1 (TRPV1) is a Ca2+ channel that is activated by capsaicin in the plasma membrane, and BPA has also been shown to activate this channel (Gonçalves et al., 2018). However, we found that ruthenium red, a TRP inhibitor, did not inhibit BPA-induced increases in NO levels (data not shown). Further studies are needed to identify the target receptors for BPA-induced Ca2+ influx.
In the present study, we found that the BPA-induced inactivation of PDI activity inhibited neurite outgrowth of PC12 cells, suggesting that S-nitrosylation of PDI inhibited neurogenesis. To our best knowledge, this is the first study to show that a µM concentrations of BPA inhibited neurite outgrowth via S-nitrosylation of PDI.
ACKNOWLEDGMENTS
We thank Dr. K. Okada for his technical support and valuable suggestions. The present study was partially supported by a JSPS KAKENHI Grant (21310045). It was also partially supported by a Grant-in-Aid from Kwansei Gakuin University.
Conflict of interest
The authors declare that there is no conflict of interest.
REFERENCES
- Agarwal, S., Yadav, A., Tiwari, S.K., Seth, B., Chauhan, L.K., Khare, P., Ray, R.S. and Chaturvedi, R.K. (2016): Dynamin-related Protein 1 Inhibition Mitigates Bisphenol A-mediated Alterations in Mitochondrial Dynamics and Neural Stem Cell Proliferation and Differentiation. J. Biol. Chem., 291, 15923-15939.
- Ahmed, R.G., Walaa, G.H. and Asmaa, F.S. (2018): Suppressive effects of neonatal bisphenol A on the neuroendocrine system. Toxicol. Ind. Health, 34, 397-407.
- Baba, K., Okada, K., Kinoshita, T. and Imaoka, S. (2009): Bisphenol A disrupts Notch signaling by inhibiting gamma-secretase activity and causes eye dysplasia of Xenopus laevis. Toxicol. Sci., 108, 344-355.
- Banker, G.A. and Cowan, W.M. (1977): Rat hippocampal neurons in dispersed cell culture. Brain Res., 126, 397-342.
- Betarbet, R., Sherer, T.B., MacKenzie, G., Garcia-Osuna, M., Panov, A.V. and Greenamyre, J.T. (2000): Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci., 3, 1301-1306.
- Bilgi, A., Abalı, R., Bilgi, P.T., Şahin, M., Tunçdemir, M. and Boran, A.B. (2019): The apoptotic effects of bisphenol A exposure on the rat ovary: an experimental study. Environ. Sci. Pollut. Res. Int., 26, 10198-10203.
- Bowman, R.E., Hagedorn, J., Madden, E. and Frankfurt, M. (2019): Effects of adolescent Bisphenol-A exposure on memory and spine density in ovariectomized female rats: adolescence vs adulthood. Horm. Behav., 107, 26-34.
- Bruno, K.A., Mathews, J.E., Yang, A.L., Frisancho, J.A., Scott, A.J., Greyner, H.D., Molina, F.A., Greenaway, M.S., Cooper, G.M., Bucek, A., Morales-Lara, A.C., Hill, A.R., Mease, A.A., Di Florio, D.N., Sousou, J.M., Coronado, A.C., Stafford, A.R. and Fairweather, D. (2019): BPA Alters Estrogen Receptor Expression in the Heart After Viral Infection Activating Cardiac Mast Cells and T Cells Leading to Perimyocarditis and Fibrosis. Front. Endocrinol. (Lausanne), 10, 598.
- Charisiadis, P., Andrianou, X.D., van der Meer, T.P., den Dunnen, W.F., Swaab, D.F., Wolffenbuttel, B.H., Makris, K.C. and van Vliet-Ostaptchouk, J.V. (2018): Possible Obesogenic Effects of Bisphenols Accumulation in the Human Brain. Sci. Rep., 8, 8186.
- Cho, J.H., Kim, A.H., Lee, S., Lee, Y., Lee, W.J., Chang, S.C. and Lee, J. (2018): Sensitive neurotoxicity assessment of bisphenol A using double immunocytochemistry of DCX and MAP2. Arch. Pharm. Res., 41, 1098-1107.
- Chouhan, S., Yadav, S.K., Prakash, J., Westfall, S., Ghosh, A., Agarwal, N.K. and Singh, S.P. (2015): Increase in the expression of inducible nitric oxide synthase on exposure to bisphenol A: a possible cause for decline in steroidogenesis in male mice. Environ. Toxicol. Pharmacol., 39, 405-416.
- Emmrich, J.V., Hornik, T.C., Neher, J.J. and Brown, G.C. (2013): Rotenone induces neuronal death by microglial phagocytosis of neurons. FEBS J., 280, 5030-5038.
- Feiteiro, J., Mariana, M., Glória, S. and Cairrao, E. (2018): Inhibition of L-type calcium channels by Bisphenol A in rat aorta smooth muscle. J. Toxicol. Sci., 43, 579-586.
- Fujimoto, T. and Aou, S. (2018): Prenatal bisphenol A exposure is associated with medial amygdala neuron hyperresponsiveness to predator odor in rats. J. Toxicol. Sci., 43, 531-536.
- Fujiwara, Y., Miyazaki, W., Koibuchi, N. and Katoh, T. (2018): The Effects of Low-Dose Bisphenol A and Bisphenol F on Neural Differentiation of a Fetal Brain-Derived Neural Progenitor Cell Line. Front. Endocrinol. (Lausanne), 9, 24.
- Fukushima, N. and Nagao, T. (2018): Exposure to bisphenol A affects GABAergic neuron differentiation in neurosphere cultures. Neuroreport, 29, 712-717.
- Ge, L.C., Chen, Z.J., Liu, H.Y., Zhang, K.S., Liu, H., Huang, H.B., Zhang, G., Wong, C.K., Giesy, J.P., Du, J. and Wang, H.S. (2014): Involvement of activating ERK1/2 through G protein coupled receptor 30 and estrogen receptor α/β in low doses of bisphenol A promoting growth of Sertoli TM4 cells. Toxicol. Lett., 226, 81-89.
- Gonçalves, R., Zanatta, A.P., Cavalari, F.C., do Nascimento, M.A., Delalande-Lecapitaine, C., Bouraïma-Lelong, H. and Silva, F.R. (2018): Acute effect of bisphenol A: signaling pathways on calcium influx in immature rat testes. Reprod. Toxicol., 77, 94-102.
- Görlach, A., Bertram, K., Hudecova, S. and Krizanova, O. (2015): Calcium and ROS: A mutual interplay. Redox Biol., 6, 260-271.
- Greene, L.A. and Tischler, A.S. (1976): Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. USA, 73, 2424-2428.
- Grohs, M.N., Reynolds, J.E., Liu, J., Martin, J.W., Pollock, T., Lebel, C. and Dewey, D.; APrON Study Team. (2019): Prenatal maternal and childhood bisphenol a exposure and brain structure and behavior of young children. Environ. Health, 18, 85.
- Hashimoto, S., Okada, K. and Imaoka, S. (2008): Interaction between bisphenol derivatives and protein disulphide isomerase (PDI) and inhibition of PDI functions: requirement of chemical structure for binding to PDI. J. Biochem., 144, 335-342.
- He, Y., Imam, S.Z., Dong, Z., Jankovic, J., Ali, S.F., Appel, S.H. and Le, W. (2003): Role of nitric oxide in rotenone-induced nigro-striatal injury. J. Neurochem., 86, 1338-1345.
- Hjorth, J.J., van Pelt, J., Mansvelder, H.D. and van Ooyen, A. (2014): Competitive dynamics during resource-driven neurite outgrowth. PLoS One, 9, e86741.
- Honjo, Y., Ayaki, T., Tomiyama, T., Horibe, T., Ito, H., Mori, H., Takahashi, R. and Kawakami, K. (2017): Decreased levels of PDI and P5 in oligodendrocytes in Alzheimer’s disease. Neuropathology, 37, 495-501.
- Kim, K., Son, T.G., Park, H.R., Kim, S.J., Kim, H.S., Kim, H.S., Kim, T.S., Jung, K.K., Han, S.Y. and Lee, J. (2009): Potencies of bisphenol A on the neuronal differentiation and hippocampal neurogenesis. J. Toxicol. Environ. Health A, 72, 1343-1351.
- Krug, A.K., Balmer, N.V., Matt, F., Schönenberger, F., Merhof, D. and Leist, M. (2013): Evaluation of a human neurite growth assay as specific screen for developmental neurotoxicants. Arch. Toxicol., 87, 2215-2231.
- Li, N., Ragheb, K., Lawler, G., Sturgis, J., Rajwa, B., Melendez, J.A. and Robinson, J.P. (2003): Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem., 278, 8516-8525.
- Li, X., Liu, Z., Tamashiro, K., Shi, B., Rudnicki, D.D., Ross, C.A., Moran, T.H. and Smith, W.W. (2010): Synphilin-1 exhibits trophic and protective effects against Rotenone toxicity. Neuroscience, 165, 455-462.
- Liang, X., Yin, N., Liang, S., Yang, R., Liu, S., Lu, Y., Jiang, L., Zhou, Q., Jiang, G. and Faiola, F. (2020): Bisphenol A and several derivatives exert neural toxicity in human neuron-like cells by decreasing neurite length. Food Chem. Toxicol., 135, 111015.
- Liu, G., Wang, P., Li, X., Li, Y., Xu, S., Uéda, K., Chan, P. and Yu, S. (2013): Alpha-synuclein promotes early neurite outgrowth in cultured primary neurons. J. Neural Transm. (Vienna), 120, 1331-1343.
- Luo, C., Rajput, A.H., Akhtar, S. and Rajput, A. (2007): Alpha-synuclein and tyrosine hydroxylase expression in acute rotenone toxicity. Int. J. Mol. Med., 19, 517-521.
- Matsushima, A., Liu, X., Okada, H., Shimohigashi, M. and Shimohigashi, Y. (2010): Bisphenol AF is a full agonist for the estrogen receptor ERalpha but a highly specific antagonist for ERbeta. Environ. Health Perspect., 118, 1267-1272.
- Miyake, Y., Hashimoto, S., Sasaki, Y., Kudo, T., Oguro, A. and Imaoka, S. (2014): Endoplasmic reticulum protein (ERp) 29 binds as strongly as protein disulfide isomerase (PDI) to bisphenol A. Chem. Res. Toxicol., 27, 501-506.
- Molina-Molina, J.M., Amaya, E., Grimaldi, M., Sáenz, J.M., Real, M., Fernández, M.F., Balaguer, P. and Olea, N. (2013): In vitro study on the agonistic and antagonistic activities of bisphenol-S and other bisphenol-A congeners and derivatives via nuclear receptors. Toxicol. Appl. Pharmacol., 272, 127-136.
- Nakamura, M., Yamanaka, H., Oguro, A. and Imaoka, S. (2018): Bisphenol A induces Nrf2-dependent drug-metabolizing enzymes through nitrosylation of Keap1. Drug Metab. Pharmacokinet., 33, 194-202.
- Nakamura, T. and Lipton, S.A. (2011): Redox modulation by S-nitrosylation contributes to protein misfolding, mitochondrial dynamics, and neuronal synaptic damage in neurodegenerative diseases. Cell Death Differ., 18, 1478-1486.
- Nakamura, T., Tu, S., Akhtar, M.W., Sunico, C.R., Okamoto, S. and Lipton, S.A. (2013): Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron, 78, 596-614.
- Naughton, M., McMahon, J., Healy, S. and FitzGerald, U. (2019): Profile of the unfolded protein response in rat cerebellar cortical development. J. Comp. Neurol., 527, 2910-2924.
- Negishi, M. and Katoh, H. (2002): Rho family GTPases as key regulators for neuronal network formation. J. Biochem., 132, 157-166.
- Ng, W.P., Cartel, N., Roder, J., Roach, A. and Lozano, A. (1996): Human central nervous system myelin inhibits neurite outgrowth. Brain Res., 720, 17-24.
- Noguchi, S., Nakatsuka, M., Asagiri, K., Habara, T., Takata, M., Konishi, H. and Kudo, T. (2002): Bisphenol A stimulates NO synthesis through a non-genomic estrogen receptor-mediated mechanism in mouse endothelial cells. Toxicol. Lett., 135, 95-101.
- Nomura, Y. (2004): Neuronal apoptosis and protection: effects of nitric oxide and endoplasmic reticulum-related proteins. Biol. Pharm. Bull., 27, 961-963.
- Obukuro, K., Nobunaga, M., Takigawa, M., Morioka, H., Hisatsune, A., Isohama, Y., Shimokawa, H., Tsutsui, M. and Katsuki, H. (2013): Nitric oxide mediates selective degeneration of hypothalamic orexin neurons through dysfunction of protein disulfide isomerase. J. Neurosci., 33, 12557-12568.
- Oguro, A., Inoue, T., Kudoh, S.N. and Imaoka, S. (2018): 14,15-epoxyeicosatrienoic acid produced by cytochrome P450s enhances neurite outgrowth of PC12 and rat hippocampal neuronal cells. Pharmacol. Res. Perspect., 6, e00428.
- Oka, T., Adati, N., Shinkai, T., Sakuma, K., Nishimura, T. and Kurose, K. (2003): Bisphenol A induces apoptosis in central neural cells during early development of Xenopus laevis. Biochem. Biophys. Res. Commun., 312, 877-882.
- Okada, K., Imaoka, S., Hashimoto, S., Hiroi, T. and Funae, Y. (2007): Over-expression of protein disulfide isomerase reduces the release of growth hormone induced by bisphenol A and/or T3. Mol. Cell. Endocrinol., 278, 44-51.
- Pandey, A.K. and Deshpande, S.B. (2015): Bisphenol A depresses monosynaptic and polysynaptic reflexes in neonatal rat spinal cord in vitro involving estrogen receptor-dependent NO-mediated mechanisms. Neuroscience, 289, 349-357.
- Rosenfeld, C.S. (2017): Neuroendocrine disruption in animal models due to exposure to bisphenol A analogues. Front. Neuroendocrinol., 47, 123-133.
- Sai, Y., Wu, Q., Le, W., Ye, F., Li, Y. and Dong, Z. (2008): Rotenone-induced PC12 cell toxicity is caused by oxidative stress resulting from altered dopamine metabolism. Toxicol. In Vitro, 22, 1461-1468.
- Seki, S., Aoki, M., Hosokawa, T., Saito, T., Masuma, R., Komori, M. and Kurasaki, M. (2011): Bisphenol-A suppresses neurite extension due to inhibition of phosphorylation of mitogen-activated protein kinase in PC12 cells. Chem. Biol. Interact., 194, 23-30.
- Suzuki, T., Mizuo, K., Nakazawa, H., Funae, Y., Fushiki, S., Fukushima, S., Shirai, T. and Narita, M. (2003): Prenatal and neonatal exposure to bisphenol-A enhances the central dopamine D1 receptor-mediated action in mice: enhancement of the methamphetamine-induced abuse state. Neuroscience, 117, 639-644.
- Tanaka, S., Uehara, T. and Nomura, Y. (2000): Up-regulation of protein-disulfide isomerase in response to hypoxia/brain ischemia and its protective effect against apoptotic cell death. J. Biol. Chem., 275, 10388-10393.
- Tiwari, S.K., Agarwal, S., Seth, B., Yadav, A., Ray, R.S., Mishra, V.N. and Chaturvedi, R.K. (2015): Inhibitory Effects of Bisphenol-A on Neural Stem Cells Proliferation and Differentiation in the Rat Brain Are Dependent on Wnt/β-Catenin Pathway. Mol. Neurobiol., 52, 1735-1757.
- Uehara, T., Nakamura, T., Yao, D., Shi, Z.Q., Gu, Z., Ma, Y., Masliah, E., Nomura, Y. and Lipton, S.A. (2006): S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature, 441, 513-517.
- Urriola-Muñoz, P., Lagos-Cabré, R. and Moreno, R.D. (2014): A mechanism of male germ cell apoptosis induced by bisphenol-A and nonylphenol involving ADAM17 and p38 MAPK activation. PLoS One, 9, e113793.
- Vuori, K., Myllylä, R., Pihlajaniemi, T. and Kivirikko, K.I. (1992): Expression and site-directed mutagenesis of human protein disulfide isomerase in Escherichia coli. This multifunctional polypeptide has two independently acting catalytic sites for the isomerase activity. J. Biol. Chem., 267, 7211-7214.
- Wang, H., Chang, L., Aguilar, J.S., Dong, S. and Hong, Y. (2019): Bisphenol-A exposure induced neurotoxicity in glutamatergic neurons derived from human embryonic stem cells. Environ. Int., 127, 324-332.
- Wu, X., Majumder, A., Webb, R. and Stice, S.L. (2016): High content imaging quantification of multiple in vitro human neurogenesis events after neurotoxin exposure. BMC Pharmacol. Toxicol., 17, 62.
- Wu, X.F., Wang, A.F., Chen, L., Huang, E.P., Xie, W.B., Liu, C., Huang, W.Y., Chen, C.X., Qiu, P.M. and Wang, H.J. (2014): S-Nitrosylating protein disulphide isomerase mediates α-synuclein aggregation caused by methamphetamine exposure in PC12 cells. Toxicol. Lett., 230, 19-27.
- Xiong, N., Long, X., Xiong, J., Jia, M., Chen, C., Huang, J., Ghoorah, D., Kong, X., Lin, Z. and Wang, T. (2012): Mitochondrial complex I inhibitor rotenone-induced toxicity and its potential mechanisms in Parkinson’s disease models. Crit. Rev. Toxicol., 42, 613-632.
- Xiong, Z.K., Lang, J., Xu, G., Li, H.Y., Zhang, Y., Wang, L., Su, Y. and Sun, A.J. (2015): Excessive levels of nitric oxide in rat model of Parkinson’s disease induced by rotenone. Exp. Ther. Med., 9, 553-558.
- Yin, N., Liang, X., Liang, S., Liang, S., Yang, R., Hu, B., Cheng, Z., Liu, S., Dong, H., Liu, S. and Faiola, F. (2019): Embryonic stem cell- and transcriptomics-based in vitro analyses reveal that bisphenols A, F and S have similar and very complex potential developmental toxicities. Ecotoxicol. Environ. Saf., 176, 330-338.
- Yoneda, T., Hiroi, T., Osada, M., Asada, A. and Funae, Y. (2003): Non-genomic modulation of dopamine release by bisphenol-A in PC12 cells. J. Neurochem., 87, 1499-1508.
- Zhang, H., Wang, Z., Meng, L., Kuang, H., Liu, J., Lv, X., Pang, Q. and Fan, R. (2020): Maternal exposure to environmental bisphenol A impairs the neurons in hippocampus across generations. Toxicology, 432, 152393.
- Zschätzsch, M., Oliva, C., Langen, M., De Geest, N., Ozel, M.N., Williamson, W.R., Lemon, W.C., Soldano, A., Munck, S., Hiesinger, P.R., Sanchez-Soriano, N. and Hassan, B.A. (2014): Regulation of branching dynamics by axon-intrinsic asymmetries in Tyrosine Kinase Receptor signaling. eLife, 3, e01699.