Abstract
Tumor necrosis factor receptor-associated factor 2 (TRAF2) is an essential component of tumor necrosis factor-α (TNF-α) signaling that regulates nuclear factor-κB (NF-κB) and c-Jun N-terminal kinase (JNK) pathways, and compelling evidence has demonstrated that TRAF2 suppresses TNF-α-induced cytotoxicity. On the other hand, it has been reported that oxidative stress-induced cytotoxicity is potentiated by TRAF2, indicating that TRAF2 both positively and negatively regulates stress-induced cytotoxicity in a context-specific manner. However, the causal role of TRAF2 in DNA damage response (DDR) remains to be explored. In this study, we assessed the function of TRAF2 in DDR induced by cisplatin, a representative DNA-damaging agent, and found that TRAF2 exerts pro-apoptotic activity through p53-dependent mechanisms at least in human fibrosarcoma cell line HT1080. TRAF2 deficient cells exhibit significant resistance to cell death induced by cisplatin, accompanied by the reduction of both p53 protein level and caspase-3 activation. Moreover, cisplatin-induced JNK activation was attenuated in TRAF2-deficient cells, and pharmacological inhibition of JNK signaling suppressed p53 stabilization. These results suggest that TRAF2 promotes p53-dependent apoptosis by activating the JNK signaling cascade in HT1080 cells. Thus, our data demonstrate a novel function of TRAF2 in cisplatin-induced DDR as a pro-apoptotic protein.
INTRODUCTION
Tumor necrosis factor receptor-associated factor 2 (TRAF2), a RING finger protein that belongs to the TRAF family proteins, is an essential signaling component of tumor necrosis factor receptor (TNFR), and negatively regulates TNFR-induced cell death (Xie, 2013; Yeh et al., 1997). TRAF2-deficient cells exhibit increased sensitivity to both tumor necrosis factor-α (TNF-α) -induced apoptosis and necroptosis (an alternative cell death modality), and similar properties of TRAF2 on cell death have been demonstrated in Fas or TNF-related apoptosis-inducing ligand (TRAIL) receptor signaling (Petersen et al., 2015; Noguchi et al., 2016; Yeh et al., 1997; Karl et al., 2014). The protective effects of TRAF2 against cell death are attributed to the ability to activate nuclear factor-κB (NF-κB) signaling or degrade caspase-8, an essential mediator of extrinsic apoptosis (Tada et al., 2001; Gonzalvez et al., 2012). On the other hand, TRAF2 transmits pro-apoptotic signals by activating mitogen-activated protein kinases (MAPKs), including c-Jun N-terminal kinase (JNK) and p38 MAPK, and thereby particularly promotes oxidative stress-induced cell death, including apoptosis (Shen et al., 2004; Lin et al., 2004; Chandel et al., 2001; Noguchi et al., 2005; Fujino et al., 2007). These reciprocal regulations of cell death by TRAF2 indicate the biological importance of TRAF2 as a signaling determinant that modulates sensitivity to stress-induced cell death.
Cellular DNA is constantly exposed to genotoxic stresses, and thus is inevitably damaged. DNA damage response (DDR) is an important mechanism to avoid the DNA damage accumulation that allows cancer development through its mutagenic consequences (Ou and Schumacher, 2018; Murakami et al., 2007). The tumor suppressor p53 is a master regulator of DDR, and drives cells into survival or death as outcomes of DDR. Therefore, loss of functional p53 has been established as a cause of cancer development (Ou and Schumacher, 2018). On the other hand, it has been reported that TRAF6, another member of the TRAF family proteins, participates in the DDR by mediating DNA damage-induced NF-κB activation (Hinz et al., 2010). Upon DNA damage, ataxia telangiectasia mutated (ATM) kinase, a DNA strand break sensor, translocates to cytosol, and then interacts and activates TRAF6, leading to the activation of the inhibitor of κB kinase (IKK) complex that stimulates canonical NF-κB signaling pathways. In addition, it turned out that IKK negatively regulates p53 stability through its direct phosphorylation, suggesting that TRAF6 acts as a negative regulator of p53 (Xia et al., 2009). Nevertheless, little attention has been paid to the involvement of TRAF2 in the DDR.
Cisplatin is a chemotherapeutic agent widely prescribed for the treatment of various cancers (Dasari and Tchounwou, 2014). It is well known that cisplatin causes DNA damage that leads to the stabilization of p53, and then exerts its cytotoxic effects by stimulating p53-dependent apoptotic pathways, resulting in cancer cell death (Jiang et al., 2004; Fridman and Lowe, 2003). However, a crucial issue is that human cancer cells frequently acquire resistance to cisplatin (Stewart, 2007). Meanwhile, its strong cytotoxicity frequently dysregulates diverse cellular functions in normal cells, and thereby causes a wide variety of side effects, including bone marrow suppression, deafness, renal dysfunction, and vomiting (Florea and Büsselberg, 2011). Therefore, to elucidate molecular mechanisms underlying cisplatin-induced cytotoxicity may provide therapeutic benefit to overcome the resistance and reduce risk of the adverse reactions. In the present study, we examined the potential role of TRAF2 in cisplatin-induced cell death, and found that TRAF2 promotes cisplatin-induced apoptosis by stabilizing p53 expression through the JNK activation.
MATERIALS AND METHODS
Cell Culture and reagents
Human fibrosarcoma cell line HT1080 and human embryonic kidney (HEK) 293A cells were grown in Dulbecco’s Modified Eagle Medium (DMEM), 10% heat-inactivated fetal bovine serum (FBS), and 1% penicillin-streptomycin solution, at 37°C under a 5% CO2 atmosphere. siRNAs were purchased from Qiagen (Hilden, Germany) (TRAF2 #1: SI00129619, TRAF2 #2: SI03073455) as described previously (Noguchi et al., 2016). AllStars negative control siRNA (Qiagen) was used as a control. siRNAs were transfected using Lipofectamine RNAiMAX (Merck Millipore, Burlington, VT, USA), according to the manufacturer’s protocol. All reagents were obtained from commercial sources; Dimethyl sulfoxide (DMSO), cisplatin, SP600125, U0126 (Wako, Tokyo, Japan), SB203580 (Santa Cruz, Dallas, TX, USA), Z-VAD-fmk (Peptide Institute, Osaka, Japan). The antibodies used were against caspase-3, p53, phospho-JNK (Cell Signaling, Danvers, MA, USA), α-tubulin, JNK (Santa Cruz), and β-actin (Wako). Monoclonal (used in Fig. 1D) and polyclonal (used in Fig. 1A and 1E) antibodies against TRAF2 are purchased from Becton, Dickinson and Company (Franklin Lakes, NJ, USA) and Santa Cruz, respectively.

Cell viability assay was performed as described previously (Noguchi et al., 2018). Cells were seeded on 96-well plates. After indicated stimulation, cell viability was measured by phenazine methosulfate (PMS)/3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) assay using Cell Titer 96 Cell Proliferation Assay kit (Promega, Madison, WI, USA), according to the manufacturer’s protocol. The absorbance was read at 492 nm using a microplate reader. Data are normalized to control (100%) without stimulus.
Immunoblot
Cells were lysed with DISC lysis buffer TX [20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 10% Glycerol, and 1% protease inhibitor cocktails (Nacalai Tesque, Kyoto, Japan)]. After centrifugation, the cell extracts were resolved by SDS-PAGE and analyzed as described previously (Noguchi et al., 2018). The blots were developed with ECL (Merck Millipore).
Generation of knockout cell lines
Knockout (KO) cells were generated using the CRISPR/Cas9 system as described previously (Hirata et al., 2017; Sekiguchi et al., 2019). Knockout cells were generated using the CRISPR/Cas9 system (Mali et al., 2013; Cong et al., 2013). Two guide RNAs (gRNAs) were designed to target a region in the exon 3 of p53 gene (5’- ATCTGAGCAGCGCTCATGGTGGG -3’) and that in the exon 2 of TRAF2 gene (5’- CCTGCAGAAACGTCCTCCGCAGG -3’), using CRISPRdirect (https://crispr.dbcls.jp) (Naito et al., 2015). gRNA-encoding oligonucleotide was cloned into lentiCRISPRv2 plasmid (addgene) (Sanjana et al., 2014), and the plasmid was transfected with HEK293A cells together with a packaging plasmid psPAX2 and an envelope plasmid pVSV-G. The supernatants were collected and used to infect HT1080 cells, and then infected cells were selected with puromycin and cloned by limiting dilution to obtain 100% efficiency. To determine the mutations of p53 and TRAF2 in cloned cells, genomic sequence around the target region was analyzed by PCR-direct sequencing using extracted DNA from each clone as a template and the following primers: 5’- AGAGACCCCAGTTGCAAACC -3’ and 5’- CCCTGCCCTCAACAAGATGT -3’ for p53; 5’- AGGTGTAACGTGCTGTGTGT -3’ and 5’- CGTGGCTCTAAAACCAGCCT -3’ for TRAF2.
NF-κB reporter assay
NF-κB reporter assays were performed essentially as described (Kudoh et al., 2018). Cells were transfected using Polyethylenimine “Max” (Polysciences, Warrington, PA, USA) with a plasmid mix containing a NF-κB luciferase reporter plasmid, a renilla luciferase plasmid for normalization, and an empty plasmid. After 24 hr, cells were treated with cisplatin. Firefly and renilla luciferase activities were quantified with dual luciferase reporter assay system (Promega).
RESULTS AND DISCUSSION
TRAF2 is required for cisplatin-induced apoptosis
It has been demonstrated that TRAF2 mediates both anti- and pro-apoptotic signals (Yeh et al., 1997; Noguchi et al., 2016, 2005; Petersen et al., 2015; Karl et al., 2014; Shen et al., 2004). Therefore, in order to examine whether TRAF2 is involved in cell death induced by DNA damage, we performed TRAF2 knockdown experiments using human fibrosarcoma cell line HT1080, in which sensitivity to TNF-α-induced apoptosis has been enhanced by TRAF2 knockdown (Noguchi et al., 2016). Both independent small interfering RNAs (siRNA) against TRAF2 reduced protein expression of TRAF2 in HT1080 cells (Fig. 1A). Interestingly, cell viability at 24 hr after the treatment with 20 μM cisplatin was partially but significantly recovered by TRAF2 knockdown (Fig. 1B). The reduction of cell viability caused by cisplatin was largely abrogated by co-treatment with Z-VAD-fmk, a pan-caspase inhibitor, indicating that cisplatin exerts its cytotoxicity mainly through apoptosis in HT1080 cells (Fig. 1C). An essential enzymatic function of caspase-3 as a main executor of apoptosis have been firmly established, although recent studies have demonstrated non-enzymatic functions of caspase-3 (Kim et al., 2018; Brentnall et al., 2014; Porter and Jänicke, 1999; Yokosawa et al., 2019). We thus tested whether TRAF2 is required for cisplatin-induced caspase-3 activation. As shown in Fig. 1D, we found that cisplatin-induced cleavage of caspase-3, a hallmark of caspase-3 activation, is suppressed in TRAF2 knockdown HT1080 cells. Therefore, to confirm the involvement of TRAF2 in cisplatin-induced apoptosis, we established TRAF2 KO HT1080 cells by using Clustered Regularly Interspaced Short Palindromic Repeat/CRISPR-associated protein-9 nuclease (CRISPR/Cas9) system, and checked TRAF2 expression by immunoblotting (Fig. 1E). As expected, two independently isolated TRAF2 KO HT1080 cells were significantly resistant to cisplatin-induced apoptosis, suggesting that TRAF2 positively regulates cisplatin-induced apoptosis in HT1080 cells (Fig. 1F).
TRAF2 is required for cisplatin-induced stabilization of p53
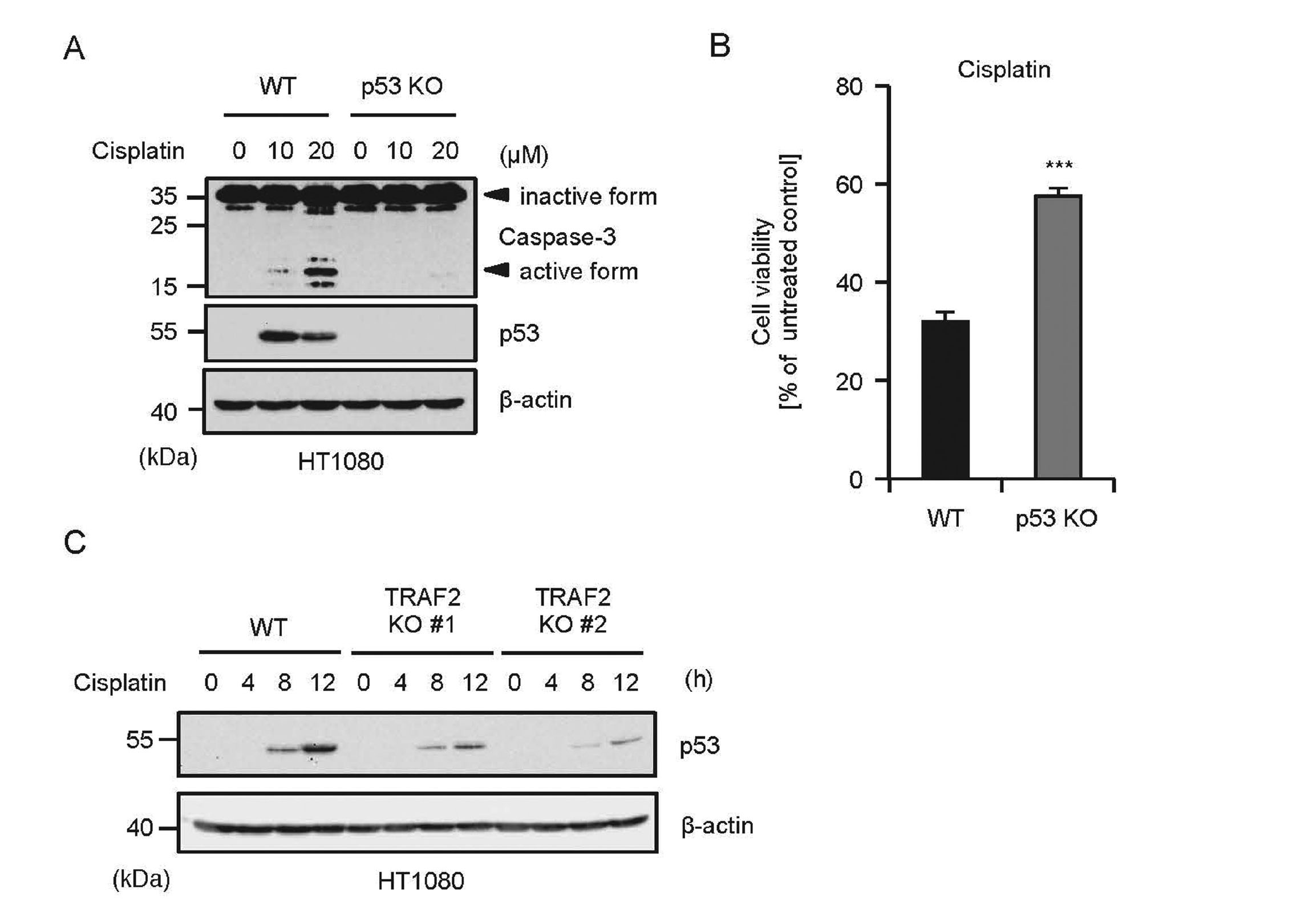
It is widely known that cisplatin exerts its pro-apoptotic activities through the p53 stabilization (Fridman and Lowe, 2003), and we observed that cisplatin induces the p53 stabilization in HT1080 cells (Fig. 2A). To confirm the requirement of p53 for cisplatin-induced apoptosis in HT1080 cells, we established p53 KO HT1080 cells. In agreement with other cell types shown in previous studies, we found that cisplatin-induced cleavage of caspase-3 is largely attenuated in p53 KO HT1080 cells (Fig. 2A) (Wei et al., 2007; He et al., 2013). Moreover, p53 KO HT1080 cells exhibited significant resistance to cisplatin-induced cell death (Fig. 2B). These findings indicate that p53-mediated caspase-3 activation plays a key role in cisplatin-induced apoptosis in HT1080 cells. Therefore, we next examined the functional links between p53 and TRAF2. Interestingly, a recent report has demonstrated that oncoprotein latent membrane protein 1 (LMP1) derived from Epstein-Barr virus enhances p53 stability through its interaction with TRAF2 (Li et al., 2012). Although the regulatory mechanism mediated by virus proteins is only observed under limited conditions, this finding raises the possibility that TRAF2 contributes to stress-induced p53 stabilization. We thus investigated the requirement of TRAF2 for cisplatin-induced p53 stabilization. As shown in Fig. 2C, cisplatin-induced p53 stabilization was clearly attenuated by knockout of TRAF2, indicating that TRAF2 is required for cisplatin-induced p53 stabilization that causes caspase-3 activation.
In TNFR signaling, TRAF2 regulates the NF-κB and JNK signaling pathways in collaboration with TRAF5 and MAPK/ERK kinase kinase-1 (MEKK1), respectively (Baud et al., 1999; Yuasa et al., 1998; Tada et al., 2001). Interestingly, JNK has been identified as a p53 kinase that phosphorylates serine 34 of p53 (Hu et al., 1997). We thus speculated that TRAF2 promotes cisplatin-induced p53 stabilization by activating the JNK signaling. As shown in Fig. 3A, pharmacological inhibition of JNK by the JNK inhibitor SP600125 clearly suppressed cisplatin-induced p53 stabilization, whereas the MAPK inhibitors for p38 MAPK (SB203580) and ERK (U0126) failed to do so. Moreover, cisplatin-induced JNK activation is partially but certainly attenuated in TRAF2 KO cells (Fig. 3B). Meanwhile, the JNK activation was not affected by p53 knockout (Fig. 3C). These findings indicate that TRAF2-mediated JNK activation is an upstream event of p53, and contributes to p53 stabilization, leading to caspase-3-mediated apoptosis. Finally, we performed luciferase reporter assays of NF-κB to examine the contribution of the TRAF2-NF-κB pathway to DDR in HT1080 cells. However, cisplatin failed to activate the reporter gene, whereas TNF-α clearly activated that (Fig. 3D). The contribution of the TRAF2-NF-κB pathway to DDR seems to be relatively small in HT1080 cells. It is not required to take into consideration the anti-apoptotic effect of TRAF2 through NF-κB activation in HT1080 cells exposed to DNA damage, and thereby, we observed the pro-apoptotic effect of TRAF2.

The TRAF2-JNK axis contributes to cell survival in the presence of TNF-α, and promotes cell death under oxidative stress condition (Yeh et al., 1997; Noguchi et al., 2005, 2008). On the basis of these results, we concluded that, at least in HT1080 cells, the TRAF2-JNK axis promotes the stabilization of p53 in response to DNA damage, which amplifies the p53-mediated pro-apoptotic signals. In this regard, it is possible that Peg3/Pw1, a pro-apoptotic protein induced by p53, is involved in the TRAF2-dependent p53 activation (Schwarzkopf et al., 2006). Interestingly, it has been reported previously that Peg3/Pw1 interacts with and activates TRAF2, suggesting that p53-mediated upregulation of Peg3/Pw1 drives a positive feedback loop for the TRAF2-JNK signaling axis (Relaix et al., 1998). Therefore, the activation or expression status of Peg3/Pw1 may determine the contribution of TRAF2 to cisplatin-induced apoptosis. Meanwhile, it has been suggested that p53-dependent apoptosis is responsible for cisplatin-induced nephrotoxicity in rats (Wei et al., 2007). Notably, the expression levels of TRAF2 are upregulated by cisplatin in rats, which exacerbates nephrotoxicity (Alhoshani et al., 2017). These observations suggest that TRAF2 enhances p53-dependent apoptosis induced by cisplatin. On the contrary, upregulation of glutathione S-transferase P1-1 (GSTP1-1) in osteosarcoma causes cisplatin resistance (Pasello et al., 2008). Interestingly, GSTP1-1 interacts with TRAF2, and inactivates TRAF2-dependent signals, suggesting that loss of the TRAF2 activation is responsible, at least in part, for GSTP1-1-driven cisplatin resistance in osteosarcoma (Wu et al., 2006; Sau et al., 2012). Collectively, these observations support our working model that TRAF2 promotes cisplatin-induced apoptosis.
Genetic alterations of TRAF2, including deletion, mutation and gene amplification, are found in various cancers (Zhu et al., 2018). In particular, the expression levels of TRAF2 are increased in prostate cancer, pancreatic cancer, lung cancer, and gastric cancer (Zhu et al., 2018). On the other hand, loss-of-function mutants of TRAF2 are observed in ovarian cancer, uterine cancer, and esophageal cancer (Zhu et al., 2018). Our findings that TRAF2 may determine the sensitivity to cisplatin-induced apoptosis indicate that cisplatin is more effective for TRAF2-overexpressing cancer. Thus, although further studies are necessary to certify the biological significance of TRAF2 in DDR, our findings reveal a novel function of TRAF2 in cisplatin-induced apoptosis, which may lead to new therapeutic strategies to overcome cisplatin resistance.
ACKNOWLEDGMENTS
We thank all members of Lab of Health Chemistry for helpful discussions. This work was supported by JSPS KAKENHI Grant Numbers JP18J20440, JP18H02567 and JP18K06622, and by MEXT KAKENHI JP17H05518 and JP19H05282. This work was also supported by the Fugaku Trust for Medicinal Research, and the Takeda Science Foundation.
Conflict of interest
The authors declare that there is no conflict of interest.
REFERENCES
- Alhoshani, A.R., Hafez, M.M., Husain, S., Al-Sheikh, A.M., Alotaibi, M.R., Al Rejaie, S.S., Alshammari, M.A., Almutairi, M.M. and Al-Shabanah, O.A. (2017): Protective effect of rutin supplementation against cisplatin-induced Nephrotoxicity in rats. BMC Nephrol., 18, 194.
- Baud, V., Liu, Z.G., Bennett, B., Suzuki, N., Xia, Y. and Karin, M. (1999): Signaling by proinflammatory cytokines: oligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes Dev., 13, 1297-1308.
- Brentnall, M., Weir, D.B., Rongvaux, A., Marcus, A.I. and Boise, L.H. (2014): Procaspase-3 regulates fibronectin secretion and influences adhesion, migration and survival independently of catalytic function. J. Cell Sci., 127, 2217-2226.
- Chandel, N.S., Schumacker, P.T. and Arch, R.H. (2001): Reactive oxygen species are downstream products of TRAF-mediated signal transduction. J. Biol. Chem., 276, 42728-42736.
- Cong, L., Ran, F.A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P.D., Wu, X., Jiang, W., Marraffini, L.A. and Zhang, F. (2013): Multiplex genome engineering using CRISPR/Cas systems. Science, 339, 819-823.
- Dasari, S. and Tchounwou, P.B. (2014): Cisplatin in cancer therapy: molecular mechanisms of action. Eur. J. Pharmacol., 740, 364-378.
- Florea, A.M. and Büsselberg, D. (2011): Cisplatin as an anti-tumor drug: cellular mechanisms of activity, drug resistance and induced side effects. Cancers (Basel), 3, 1351-1371.
- Fridman, J.S. and Lowe, S.W. (2003): Control of apoptosis by p53. Oncogene, 22, 9030-9040.
- Fujino, G., Noguchi, T., Matsuzawa, A., Yamauchi, S., Saitoh, M., Takeda, K. and Ichijo, H. (2007): Thioredoxin and TRAF family proteins regulate reactive oxygen species-dependent activation of ASK1 through reciprocal modulation of the N-terminal homophilic interaction of ASK1. Mol. Cell. Biol., 27, 8152-8163.
- Gonzalvez, F., Lawrence, D., Yang, B., Yee, S., Pitti, R., Marsters, S., Pham, V.C., Stephan, J.P., Lill, J. and Ashkenazi, A. (2012): TRAF2 Sets a threshold for extrinsic apoptosis by tagging caspase-8 with a ubiquitin shutoff timer. Mol. Cell, 48, 888-899.
- He, K., Zheng, X., Zhang, L. and Yu, J. (2013): Hsp90 inhibitors promote p53-dependent apoptosis through PUMA and Bax. Mol. Cancer Ther., 12, 2559-2568.
- Hinz, M., Stilmann, M., Arslan, S.C., Khanna, K.K., Dittmar, G. and Scheidereit, C. (2010): A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-κB activation. Mol. Cell, 40, 63-74.
- Hirata, Y., Takahashi, M., Kudoh, Y., Kano, K., Kawana, H., Makide, K., Shinoda, Y., Yabuki, Y., Fukunaga, K., Aoki, J., Noguchi, T. and Matsuzawa, A. (2017): trans-Fatty acids promote proinflammatory signaling and cell death by stimulating the apoptosis signal-regulating kinase 1 (ASK1)-p38 pathway. J. Biol. Chem., 292, 8174-8185.
- Hu, M.C., Qiu, W.R. and Wang, Y.P. (1997): JNK1, JNK2 and JNK3 are p53 N-terminal serine 34 kinases. Oncogene, 15, 2277-2287.
- Jiang, M., Yi, X., Hsu, S., Wang, C.Y. and Dong, Z. (2004): Role of p53 in cisplatin-induced tubular cell apoptosis: dependence on p53 transcriptional activity. Am. J. Physiol. Renal Physiol., 287, F1140-F1147.
- Karl, I., Jossberger-Werner, M., Schmidt, N., Horn, S., Goebeler, M., Leverkus, M., Wajant, H. and Giner, T. (2014): TRAF2 inhibits TRAIL- and CD95L-induced apoptosis and necroptosis. Cell Death Dis., 5, e1444.
- Kim, J.S., Ha, J.Y., Yang, S.J. and Son, J.H. (2018): A Novel Non-Apoptotic Role of Procaspase-3 in the Regulation of Mitochondrial Biogenesis Activators. J. Cell. Biochem., 119, 347-357.
- Kudoh, Y., Noguchi, T., Ishii, C., Maeda, K., Nishidate, A., Hirata, Y. and Matsuzawa, A. (2018): Antibiotic Vancomycin Promotes the Gene Expression of NOD-Like Receptor Families in Macrophages. BPB Reports, 1, 6-10.
- Li, L., Li, W., Xiao, L., Xu, J., Chen, X., Tang, M., Dong, Z., Tao, Q. and Cao, Y. (2012): Viral oncoprotein LMP1 disrupts p53-induced cell cycle arrest and apoptosis through modulating K63-linked ubiquitination of p53. Cell Cycle, 11, 2327-2336.
- Lin, Y., Choksi, S., Shen, H.M., Yang, Q.F., Hur, G.M., Kim, Y.S., Tran, J.H., Nedospasov, S.A. and Liu, Z.G. (2004): Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J. Biol. Chem., 279, 10822-10828.
- Mali, P., Yang, L., Esvelt, K.M., Aach, J., Guell, M., DiCarlo, J.E., Norville, J.E. and Church, G.M. (2013): RNA-guided human genome engineering via Cas9. Science, 339, 823-826.
- Murakami, S., Noguchi, T., Takeda, K. and Ichijo, H. (2007): Stress signaling in cancer. Cancer Sci., 98, 1521-1527.
- Naito, Y., Hino, K., Bono, H. and Ui-Tei, K. (2015): CRISPRdirect: software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics, 31, 1120-1123.
- Noguchi, T. (2008): ROS-dependent activation of ASKI in inflammatory signaling. J. Oral Biosci., 50, 107-114.
- Noguchi, T., Suzuki, M., Mutoh, N., Hirata, Y., Tsuchida, M., Miyagawa, S., Hwang, G.W., Aoki, J. and Matsuzawa, A. (2018): Nuclear-accumulated SQSTM1/p62-based ALIS act as microdomains sensing cellular stresses and triggering oxidative stress-induced parthanatos. Cell Death Dis., 9, 1193.
- Noguchi, T., Takeda, K., Matsuzawa, A., Saegusa, K., Nakano, H., Gohda, J., Inoue, J. and Ichijo, H. (2005): Recruitment of tumor necrosis factor receptor-associated factor family proteins to apoptosis signal-regulating kinase 1 signalosome is essential for oxidative stress-induced cell death. J. Biol. Chem., 280, 37033-37040.
- Noguchi, T., Tsuchida, M., Kogue, Y., Spadini, C., Hirata, Y. and Matsuzawa, A. (2016): Brefeldin A-Inhibited Guanine Nucleotide-Exchange Factor 1 (BIG1) Governs the Recruitment of Tumor Necrosis Factor Receptor-Associated Factor 2 (TRAF2) to Tumor Necrosis Factor Receptor 1 (TNFR1). Signaling Complexes. Int. J. Mol. Sci., 17, 1869.
- Ou, H.L. and Schumacher, B. (2018): DNA damage responses and p53 in the aging process. Blood, 131, 488-495.
- Pasello, M., Michelacci, F., Scionti, I., Hattinger, C.M., Zuntini, M., Caccuri, A.M., Scotlandi, K., Picci, P. and Serra, M. (2008): Overcoming glutathione S-transferase P1-related cisplatin resistance in osteosarcoma. Cancer Res., 68, 6661-6668.
- Petersen, S.L., Chen, T.T., Lawrence, D.A., Marsters, S.A., Gonzalvez, F. and Ashkenazi, A. (2015): TRAF2 is a biologically important necroptosis suppressor. Cell Death Differ., 22, 1846-1857.
- Porter, A.G. and Jänicke, R.U. (1999): Emerging roles of caspase-3 in apoptosis. Cell Death Differ., 6, 99-104.
- Relaix, F., Wei, X.-J., Wu, X. and Sassoon, D.A. (1998): Peg3/Pw1 is an imprinted gene involved in the TNF-NFκB signal transduction pathway. Nat. Genet., 18, 287-291.
- Sanjana, N.E., Shalem, O. and Zhang, F. (2014): Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods, 11, 783-784.
- Sau, A., Filomeni, G., Pezzola, S., D’Aguanno, S., Tregno, F.P., Urbani, A., Serra, M., Pasello, M., Picci, P., Federici, G. and Caccuri, A.M. (2012): Targeting GSTP1-1 induces JNK activation and leads to apoptosis in cisplatin-sensitive and -resistant human osteosarcoma cell lines. Mol. Biosyst., 8, 994-1006.
- Schwarzkopf, M., Coletti, D., Sassoon, D. and Marazzi, G. (2006): Muscle cachexia is regulated by a p53-PW1/Peg3-dependent pathway. Genes Dev., 20, 3440-3452.
- Sekiguchi, Y., Yamada, M., Noguchi, T., Noomote, C., Tsuchida, M., Kudoh, Y., Hirata, Y. and Matsuzawa, A. (2019): The anti-cancer drug gefitinib accelerates Fas-mediated apoptosis by enhancing caspase-8 activation in cancer cells. J. Toxicol. Sci., 44, 435-440.
- Shen, H.M., Lin, Y., Choksi, S., Tran, J., Jin, T., Chang, L., Karin, M., Zhang, J. and Liu, Z.G. (2004): Essential roles of receptor-interacting protein and TRAF2 in oxidative stress-induced cell death. Mol. Cell. Biol., 24, 5914-5922.
- Stewart, D.J. (2007): Mechanisms of resistance to cisplatin and carboplatin. Crit. Rev. Oncol. Hematol., 63, 12-31.
- Tada, K., Okazaki, T., Sakon, S., Kobarai, T., Kurosawa, K., Yamaoka, S., Hashimoto, H., Mak, T.W., Yagita, H., Okumura, K., Yeh, W.C. and Nakano, H. (2001): Critical roles of TRAF2 and TRAF5 in tumor necrosis factor-induced NF-kappa B activation and protection from cell death. J. Biol. Chem., 276, 36530-36534.
- Wei, Q., Dong, G., Yang, T., Megyesi, J., Price, P.M. and Dong, Z. (2007): Activation and involvement of p53 in cisplatin-induced nephrotoxicity. Am. J. Physiol. Renal Physiol., 293, F1282-F1291.
- Wu, Y., Fan, Y., Xue, B., Luo, L., Shen, J., Zhang, S., Jiang, Y. and Yin, Z. (2006): Human glutathione S-transferase P1-1 interacts with TRAF2 and regulates TRAF2-ASK1 signals. Oncogene, 25, 5787-5800.
- Xia, Y., Padre, R.C., De Mendoza, T.H., Bottero, V., Tergaonkar, V.B. and Verma, I.M. (2009): Phosphorylation of p53 by IkappaB kinase 2 promotes its degradation by beta-TrCP. Proc. Natl. Acad. Sci. USA, 106, 2629-2634.
- Xie, P. (2013): TRAF molecules in cell signaling and in human diseases. J. Mol. Signal., 8, 7.
- Yeh, W.C., Shahinian, A., Speiser, D., Kraunus, J., Billia, F., Wakeham, A., de la Pompa, J.L., Ferrick, D., Hum, B., Iscove, N., Ohashi, P., Rothe, M., Goeddel, D.V. and Mak, T.W. (1997): Early lethality, functional NF-kappaB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity, 7, 715-725.
- Yokosawa, T., Yamada, M., Noguchi, T., Suzuki, S., Hirata, Y. and Matsuzawa, A. (2019): Pro-caspase-3 protects cells from polymyxin B-induced cytotoxicity by preventing ROS accumulation. J. Antibiot. (Tokyo), 72, 848-852.
- Yuasa, T., Ohno, S., Kehrl, J.H. and Kyriakis, J.M. (1998): Tumor necrosis factor signaling to stress-activated protein kinase (SAPK)/Jun NH2-terminal kinase (JNK) and p38. Germinal center kinase couples TRAF2 to mitogen-activated protein kinase/ERK kinase kinase 1 and SAPK while receptor interacting protein associates with a mitogen-activated protein kinase kinase kinase upstream of MKK6 and p38. J. Biol. Chem., 273, 22681-22692.
- Zhu, S., Jin, J., Gokhale, S., Lu, A.M., Shan, H., Feng, J. and Xie, P. (2018): Genetic Alterations of TRAF Proteins in Human Cancers. Front. Immunol., 9, 2111.