Abstract
Acute exposure to hydrogen sulfide (H2S) can cause fatal acute lung injury (ALI). However, the mechanisms of H2S-induced ALI are still not fully understood. This study aims to investigate the role of the tight junction protein claudin-5 in H2S-induced ALI. In our study, Sprague-Dawley (SD) rats were exposed to H2S to establish the ALI model, and in parallel, human pulmonary microvascular endothelial cells (HPMECs) were incubated with NaHS (a H2S donor) to establish a cell model. Lung immunohistochemistry and electron microscopy assays were used to identify H2S-induced ALI, and the expression of claudin-5, p-AKT/t-AKT and p-FoxO1/t-FoxO1 was detected. Our results show that H2S promoted the formation of ALI by morphological investigation and decreased claudin-5 expression. Dexamethasone (Dex) could partly attenuate NaHS-mediated claudin-5 downregulation, and the protective effects of Dex could be partially blocked by LY294002, a PI3K/AKT/FoxO1 pathway antagonist. Moreover, as a consequence of the altered phosphorylation of AKT and FoxO1, a change in claudin-5 with the same trend was observed. Therefore, the tight junction protein claudin-5 might be considered a therapeutic target for the treatment of ALI induced by H2S and other hazardous gases.
INTRODUCTION
Hydrogen sulfide (H2S) is a colorless and toxic asphyxiating gas. H2S exposure can cause a variety of respiratory symptoms, from irritant inflammation of the nasopharynx to acute lung injury (ALI). (Mehta et al., 2014) Respiratory failure caused by ALI is the main cause of death from H2S poisoning. (Jin et al., 2013) Early, adequate, and repeated use of glucocorticoids can effectively alleviate ALI, and its efficacy has been affirmed. However, the mechanisms of H2S-induced ALI and the glucocorticoid-mediated protective effects are still not fully clear.
It is well known that the vascular endothelium plays a key role in regulating tissue fluid homeostasis and the inflammatory response. (Nomura et al., 2014) The integrity of the vascular endothelium is the basis of its barrier function. Intercellular tight junctions (TJs) function as barriers by controlling the paracellular pathway, which is composed of occludin, claudin and Jam (adhesive connexin). (Wilmes et al., 2014) It has been confirmed that the human claudin family has at least 27 members. (Whitsett and Alenghat, 2015) Claudin-5 is mainly expressed in the vascular endothelium and lung microcirculation. (Mitchell et al., 2015) Related animal and cell experiments showed that claudin-5 could increase the barrier function of endothelial cells and protect the endothelium. (Li et al., 2014) In the pathogenesis of ALI, the increase in microvascular permeability and the destruction of the blood-gas barrier are related to the downregulation of claudin-5. (Kage et al., 2014; Li et al., 2014).
Phosphatidylinositol 3-kinase (PI3K) and its downstream molecule protein kinase B (PKB/AKT) are involved in the regulation of various cellular functions, such as proliferation, differentiation, apoptosis and glucose transport. The forkhead box (Fox) protein family is composed of transcription factors with a wing-like helical structure in the DNA binding region (Jang et al., 2011). The protein family is divided into 17 subfamilies, and FoxO is a subgroup of the Fox family. As a member of the FoxO subfamily, FoxO1 contains three highly conserved phosphorylation sites (Thr24, Ser256 and Ser319). Its upstream is regulated by the PI3K/AKT phosphorylation cascade, and its phosphorylation/dephosphorylation state is closely related to transcriptional regulation. (Ward et al., 2015) FoxO1 plays an important role in regulating glycolipid metabolism, antioxidative stress, cell cycle and DNA damage repair. (Aslam et al., 2012) In addition to their involvement in the regulation of apoptosis, these factors also mediate cell-to-extracellular matrix adhesion, regulate the expression of genes involved in proinflammatory formation and thrombosis, and participate in cell repair, proliferation, and angiogenesis.
Kage et al. (Kage et al., 2014; Li et al., 2014) found that VE-cadherin can activate AKT/FoxO1 sequentially through the PI3K pathway and that FoxO1 is translocated from the nucleus to the cytoplasm after phosphorylation, thereby releasing its inhibitory effect on claudin-5 and upregulating the expression of claudin-5. Therefore, the objective of the present study was to verify whether claudin-5 is involved in H2S-induced ALI and to assess the signaling pathways involved in claudin-5 expression. In an in vivo study, Sprague-Dawley (SD) rats were exposed to H2S at concentrations that were likely to be encountered and might contribute to ALI. The damage to pulmonary alveoli and tight junctions was then evaluated by electron microscopy. In an in vitro study, we incubated human pulmonary microvascular endothelial cells (HPMECs) with NaHS (H2S donor), and then the expression of claudin-5, p-AKT/t-AKT, and p-FoxO1/t-FoxO1 was examined. Finally, the permeability of HPMECs to sodium fluorescein and FITC-dextran was detected, which would help us further understand the effect of claudin-5 on lung vascular endothelial permeability.
MATERIALS AND METHODS
Chemicals and antibodies
NaHS (#161527) and Dex (#4902) were purchased from Sigma-Aldrich (St. Louis, Missouri, USA). Gas cylinders containing 1% (10,000 ppm) H2S in nitrogen were purchased from Shangyuan GASES (Nanjing, China). A digital H2S gas analyzer was purchased from Lasting Star Safety Equipment Company (Nanjing, China). Claudin-5 antibodies were obtained from Sigma-Aldrich. All other antibodies were from Cell Signaling Technology (Danvers, MA, USA) unless otherwise stated.
Animals
Male SD rats weighing 200 ± 20 g were purchased from the Animal Center of Jiangsu Province, Nanjing, China (SCXK(Su) 2002-0031) and had free access to standard rat chow and tap water. All animals were housed in independent ventilation cages at an ambient temperature of 22-24°C and humidity of 50-60% with a 12 hr light/dark cycle.
Cell culture
HPMECs were purchased from American Type Culture Collection (ATCC), inoculated in a 25 cm2 Corning flask at a density of 4*105 cells/cm2 using specific medium for endothelial cells (ECM) containing 5% fetal bovine serum, 1% FBS endothelial growth factor and 1% penicillin/streptomycin and cultured in a cell culture incubator at 37°C and 5% CO2. Trypsin-EDTA solution was used for trypsinization and subculture of the cells.
Exposure of rats to H2S
Thirty rats were randomly divided into a control (unexposed) group and four time-point groups (n = 6 per group) using a computer-generated randomization schedule. Control rats were kept in room air, whereas the other 24 rats were exposed to 300 ppm H2S for 3 hr, returned to room air and then anesthetized by intraperitoneal administration of pentobarbital sodium at 0, 6, 12, and 24 hr after H2S exposure.
Transmission electron microscopy
The right lower lung lobe from each rat was harvested and fixed in 2.5% glutaraldehyde for 24 hr and embedded in paraffin. Small blocks of lung tissue were postfixed in 1% osmium tetroxide in PBS, dehydrated in a graded series of ethanol washes, and embedded. Ultrathin sections stained with uranylacetate and lead citrate were examined using a transmission electron microscope (JEM 1010; JEOL, Tokyo, Japan).
Immunohistochemical Examination
Three millimeter-thick serial sections of paraffin-embedded tissue were prepared by deparaffinizing, rehydrating and quenching endogenous peroxidase. Antigen retrieval was achieved using 0.05% protease XIV at 37°C for 5 min. Each section was incubated with rabbit monoclonal claudin-5 (1:100) for 1 hr at room temperature and overnight at 4°C. Following the reaction with anti-rabbit IgG (1:50,000, Jackson Immuno Research Laboratories, Baltimore, MD, USA) for 15 min, the sections were treated with aminoethyl carbazole and counterstained with Mayer’s hematoxylin. Images of immunohistochemical examination were processed by a Nikon Eclipse 80i microscope with NIS Elements software (Media Cybernetics, Silver Spring, MD, USA).
RNA extraction and real-time PCR (Q-PCR) analysis
Total RNA was isolated using RNAiso Plus (TaKaRa, Kyoto, Japan) according to the manufacturer’s instructions. RNA was dissolved in RNase-free water, and concentrations were assessed by a NANO drop ND-1000 Spectrophotometer (Nano Drop Technologies, Wilmington, DE, USA). Then, mRNA was reverse transcribed into cDNA using an RT-PCR kit (TaKaRa). The claudin-5 target sequence used was as follows: 5´-GCTTCTGGCACTCTTTGTTACCTTG-3´. The β-ACTIN target sequence was 5´-GAGACCTTCAACACCCCAGC-3´. Real-time PCR was carried out on an ABI Prism 7300 HT Sequence Detection System with a SYBR Premix Ex Taq kit (TaKaRa) in a 20 mL reaction mixture. Fold changes of the target genes were calculated using the 2-ΔΔCt method.
Cell toxicity model
HPMECs were stabilized with serum-free ECM medium. As a donor of H2S, NaHS was dissolved in sterile PBS at a concentration of 100 mmol/L of mother liquor. Fresh NaHS mother solution was added to serum-free medium to a final concentration of 500 µmol/L NaHS (Eghbal et al., 2004; Truong et al., 2006) and then incubated with HPMECs for 30 min, 1 hr, 3 hr, 6 hr, 12 hr and 24 hr.
Dex and/or LY294002 intervention model
According to a previous report (Eghbal et al., 2004; Truong et al., 2006), the in vivo intervention was as follows: Dex (2 mg/kg/day) was intraperitoneally injected for three consecutive days prior to H2S exposure. In vitro, HPMECs were incubated with Dex (100 nmol/L) in serum-free ECM for 24 hr (Prota et al., 2012), and then the medium was changed. The cells were then incubated with NaHS (500 µmol/L). According to the instruction manual, the intervention method for LY294002 in vitro was as follows: HPMECs were incubated with LY294002 (10 µmol/L) for 1 hr before NaHS treatment.
Western blot analysis
Cell lysates were prepared using RIPA buffer (Sigma, Shanghai, China) with Protease Inhibitor Cocktail (Sigma) and incubated for 30 min at 4°C. Lysates were centrifuged at 12,000 rpm at 4°C for 15 min. Then, the supernatants were collected, and protein concentrations were determined using Pierce BCA protein assay reagent (Thermo Scientific, Waltham, MA, USA). Samples with equal amounts of protein were loaded onto 10% sodium dodecyl sulfate-polyacrylamide gels, subjected to electrophoresis and subsequently blotted onto 0.22 mmol/L polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA). After blocking with 5% nonfat milk, the membranes were incubated with anti-claudin-5 (1:1000), anti-p-AKT (1:1000), anti-t-AKT (1:1000), anti-p-FoxO1 (1:1000), anti-t-FoxO1 (1:1000), or mouse anti-actin (1:5000) for at least 8 hr. After incubation with horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-goat secondary antibody (1:50,000; Jackson Immuno Research Laboratories) for 1 hr at room temperature, labeled proteins were visualized using Pierce ECL Western Blotting Substrate (Thermo Scientific). Band density was normalized to β-actin in each sample.
HPMEC permeability examination
HPMECs were seeded inside the insert (diameter, 6.5 mm; pore size, 0.4 µm; Corning; Tewksbury, MA, USA) at a density of 50,000 cells per insert well and cultured for 7 days to allow the growth of a confluent monolayer. The TEER (Ωcm2) was measured daily using a Millicell-electrical resistance system (ERS) (Millipore Co. Ltd) until its absolute value reached 300 Ωcm2) (Kage et al., 2014; Li et al., 2014). Monolayers were pretreated with or without Dex (100 nmol/L) for 24 hr and then stimulated with NaHS (500 µmol/L) for an additional 12 hr. LY294002 (10 μmol/L) was also added to the upper compartment 1 hr before NaHS treatment. Subsequently, all media were removed, and 0.6 mL of phosphate-buffered saline was added to the lower chamber. Then, 10 μL of 2 mg/mL sodium fluorescein (376 Da) or 10 mg/mL FITC-dextran (2,000 kDa) was added to the upper chamber for a 1 hr incubation. The transwell insert was then removed, and 100 µL of medium was collected. Fluorescence intensity was analyzed with a fluorescence microplate reader by using excitation and emission wavelengths of 490 and 525 nm, respectively. Calculation of the permeability coefficients was accomplished according to the clearance principle as previously published (Kage et al., 2014; Li et al., 2014). Briefly, the permeability coefficient p was calculated from the following equation: P=ΔCL*VL/CU*Δt*A(1), where ΔCL is the fluorescence intensity change in the lower chamber, CU is the initial fluorescence intensity of the medium in the upper chamber, VL is the volume of the well (1 cm3), A is the area of the membrane (0.33 cm2) that allows fluorescent tracers to permeate from the culture insert to the well plate, and Δt is the assay time (3600 sec). Last, the permeability of HPMECs, Pe, was calculated from the following equation, where Pb is the permeability of the cell culture insert without cells: 1/Pe=1/P – 1/Pb (2).
Statistical analysis
Data are expressed as the mean ± SEM for all experiments. Statistically significant differences between the treatment groups and the control group were determined by one-way ANOVA and LSD multiple comparison procedure or Student’s t-test. All graphs were generated using GraphPad Prism 8.0 (GraphPad, San Diego, CA, USA). p < 0.05 was considered statistically significant.
RESULTS
Ultrastructure of lung tissue exposed to H2S under electron microscopy
Marked H2S-induced ultrastructural abnormalities in type II lung alveolar epithelial cells were observed. The untreated cells showed no swelling of mitochondria, clear nuclei and onion-like lamellar bodies (Fig. 1A). Mitochondria showed marked edema accompanied by disintegration of lamellar bodies (Fig. 1B) and shrinking cell bodies, nuclear pyrosis, and nuclear fragmentation (Fig. 1C). In addition, the ultrastructure of the tight junction of the alveolar epithelium also showed injurious changes, from normal clear and coherent linear structure to light staining, blurred structure, loss of continuous structure and interrupted rupture (Fig. 1D-F). Lung histopathology confirmed acute lung injury, and tight junctions were also damaged after H2S exposure.
H2S regulated claudin-5 expression in lung tissues
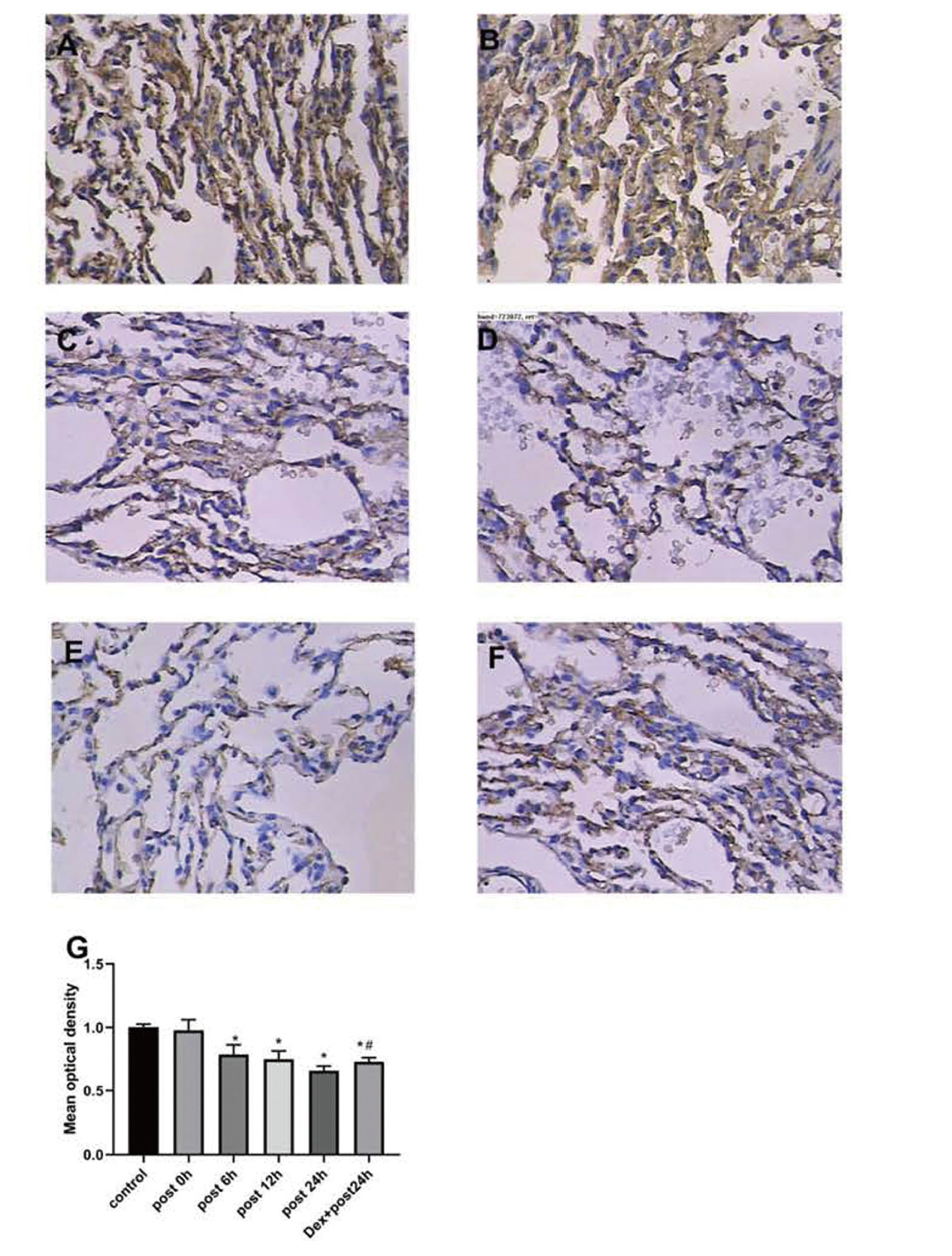
To assess the time-dependent effect of H2S, claudin-5 mRNA was detected at 0, 6, 12 and 24 hr after exposure. As shown in Fig. 2, the claudin-5 mRNA level decreased continuously from 6 hr to 24 hr after H2S exposure. To corroborate the findings that H2S downregulated claudin-5 mRNA expression in lung tissue, parallel experiments were carried out to detect the expression of claudin-5 protein in vivo. In line with the mRNA expression results, the immunohistochemical results also revealed a marked decrease in claudin-5 protein from 6 hr to 24 hr after exposure (Fig. 3). Interestingly, claudin-5 protein expression was significantly higher in the Dex+post 24 hr group (0.73 relative to control) than in the post 24 hr group (0.66 relative to control). The above in vivo experiments revealed that H2S downregulated the expression of claudin-5 at both the gene and protein levels. Dex could significantly reduce the decline in claudin-5 induced by H2S.

It was reported that the regulation of claudin-5 expression was mainly mediated by the extracellularly regulated protein PI3K/AKT/FoxO1 signaling pathway (Jang et al., 2011). Therefore, we first assessed the effects of H2S on AKT and FoxO1 phosphorylation. After 500 µmol/L NaHS stimulation, p-AKT displayed a transient increase and then a sustained significant decline from 6 hr to 24 hr after exposure (Fig. 4A, B). A similar tendency was also observed in p-FoxO1 expression. There was no significant change in the t-AKT or t-FoxO1 protein level (Fig. 4C, D). Overall, H2S decreased claudin-5 expression, which was accompanied by decreased p-AKT and p-FoxO1 levels. The results revealed that the PI3K/AKT/FoxO1 signaling pathway was activated by NaHS.
The PI3K/AKT/FoxO1 pathway participates in the claudin-5 regulation of H2S
To further explore whether the PI3K/AKT/FoxO1 pathway was implicated in the claudin-5 regulation of H2S, according to the above results, the expression of claudin-5, p-AKT/t-AKT and p-FoxO1/t-FoxO1 all decreased significantly at the 12 hr time point after exposure. These proteins were detected following Dex and/or LY294002 (a PI3K inhibitor) treatment after 12 hr exposure. As indicated in Fig. 5, the expression of claudin-5 in group Dex+post 12 hr (78% relative to control) was significantly higher than that in group post 12 hr (38% relative to control) and group Dex+post 12 hr+LY (49% relative to control). The following experiments indicated that the change trends of p-AKT/t-AKT and p-FoxO1/t-FoxO1 were similar to that of claudin-5 in the Dex+post 12 hr, post 12 hr and Dex+post 12 hr+LY groups (Figs. 6, 7). Overall, our data revealed that NaHS-mediated downregulation of claudin-5 could be partially attenuated by Dex and that the Dex-induced upregulation of claudin-5 could be markedly inhibited by LY294002, which revealed the role of the PI3K/AKT/FoxO1 pathway in claudin-5 regulation of H2S. Moreover, the phosphorylation status of these transcription factors was consistent with the observed claudin-5 alteration.



The key role of claudin-5 in pulmonary capillary permeability is well documented (Zinter et al., 2019). Therefore, it was reasonable to assay H2S-induced endothelial barrier dysfunction, as well as intervention with Dex and LY294002. HPMECs were incubated with NaHS (500 µmol/L) alone, pretreated with Dex (100 nmol/L) 24 hr ahead of NaHS treatment, or prophylactically treated with Dexand LY294002 (10 μmol/L) 24 hr and 1 hr ahead of NaHS treatment, respectively. Then, the permeability values were measured by the monolayer flux of sodium fluorescein (376 Da) or FITC-dextran (2,000 kDa) after incubation for 12 hr. As indicated in Fig. 8A, the sodium fluorescein permeability in the post-12 hr group increased significantly, reaching 2.4 times that of the control group. After the Dex intervention, the high permeability was significantly improved, reduced to 1.3 times that of the control group. However, after the combined intervention with Dex and LY294002, the permeability in the Dex+LY+post 12 hr group reached 2 times that of the control group (p < 0.05). Notably, there was no significant change in permeability in any groups measured by monolayer FITC-dextran, as indicated in Fig. 8B (p > 0.05). Our data indicated that NaHS significantly induced endothelial barrier dysfunction, Dex could partly attenuate NaHS-mediated EC barrier dysfunction as measured by the monolayer flux of sodium fluorescein, and the role of Dex could be significantly inhibited by LY294002.

DISCUSSION
The underlying cause of ALI is injury to vascular endothelial cells caused by various traumatic factors (Zinter et al., 2019). In the normal state, endothelial cells are cobblestone-like, arranged evenly and densely in a monolayer, maintaining the integrity of the endothelial barrier. Pulmonary microvascular endothelial cells regulate the permeability of the vascular barrier under physiological and pathological conditions (Millar et al., 2016) and act as the first barrier for inflammatory cells to infiltrate from vessels to alveoli during the development of ALI (Zhang et al., 2016).
The barrier function of the vascular endothelium depends on the TJ-mediated paracellular pathway (Suzuki et al., 2015). Moreover, claudin family transmembrane proteins directly regulate TJ’s permeability. Claudins are 22-27 kDa and contain four transmembrane domains and two extracellular loops (ELs). EL1 is approximately 50 amino acids long and is responsible for the selective permeability of TJs (Koval, 2013). Increased lung vascular permeability has been associated with decreased claudin-5 levels in several studies, suggesting that decreased claudin-5 has a deleterious effect on lung vascular tight junctions. Chen used 5 µmol/L simvastatin to treat pulmonary artery endothelial cells and found that claudin-5 levels increased after 16 hr, while silencing of claudin-5 significantly attenuated simvastatin-mediated EC barrier protection in response to thrombin (Chen et al., 2014). Similar studies have shown that claudin-5 downregulation is closely related to an impaired blood-gas barrier during ALI (Li et al., 2009; Xia et al., 2010).
In recent years, besides oxidation reaction and inflammatory reaction, some new mechanisms of H2S-induced ALI have been reported as well as mitochondrial damage, downregulation of sodium channels in lung epithelial cells (α-ENaC) (Jiang et al., 2014, 2015), matrix metalloproteinases-2 and 9 (MMP-2 and MMP-9) upregulation (Wang et al., 2014) and aquaporins-5 (AQP5) downregulation (Xu et al., 2017). Herein, we speculated that H2S might exert its effects on claudin-5. In the in vivo experiments, a significant reduction in claudin-5 at the gene and protein levels was observed after exposure, accompanied by the formation of ALI and damage to tight junctions in type II lung alveolar epithelial cells. In parallel, similar results were observed in an in vitro study when HPMECs were incubated with NaHS for 12 hr. These results indicated that claudin-5 might be a potential target for H2S.
Dex is widely used in the clinical treatment of ALI. Due to various causes of ALI, glucocorticoids are currently controversial in the treatment of ALI. However, it is effective for toxic gas-induced ALI, although its therapeutic mechanism is not clear at present. In vivo and in vitro experiments have confirmed that glucocorticoids enhance the barrier function of cerebral vascular endothelial cells and then reduce brain edema (Weksler et al., 2005). Therefore, we assessed claudin-5 expression affected by Dex. Our results revealed that Dex significantly attenuated the H2S-mediated downregulation of claudin-5.
Several pathways participated in the regulation of claudin-5 expression, such as JAK-STAT6-FoxO1, (Dalmasso et al., 2014) SOX18, (Gross et al., 2014) p38 MAPK/NF-κB and PI3K-AKT-FoxO1. Of all these pathways, the enhancement of AKT/FoxO1 phosphorylation was reported to be associated with beneficial effects in ALI (Jang et al., 2011). Therefore, we detected the expression of p-AKT/t-AKT and p-FoxO1/t-FoxO1 in the NaHS-exposed group and the Dex intervention group. As we hypothesized, the PI3K/AKT/FoxO1 signaling pathway is involved in the negative regulation of H2S on claudin-5, including the positive regulation of Dex on claudin-5. Consistent with this mechanism, treatment with LY294002 diminished AKT and FoxO1 phosphorylation induced by Dex and decreased claudin-5 levels. The result was a transient increase in p-AKT and p-FOXO1 in the early stage of H2S treatment, which was associated with the individual moderate stress reaction (Jang et al., 2011).
PI3K/AKT has been implicated in diseases ranging from cancer and diabetes to neurodegeneration, as well as being of functional relevance in anti-inflammatory and anti-oxidative signaling (Crossland et al., 2010). A previous study (Dalmasso et al., 2014) showed that VE-cadherin upregulated claudin-5 indirectly by releasing the inhibitory effect of FoxO1 through the induction of AKT. Chen et al. (Li et al., 2009) reported that membrane translocation of VE-cadherinin response to simvastatin and increased phosphorylation of FoxO1 is consistent with the observed increase in claudin-5 expression as the downregulation of FoxO1. As our experiment was not designed to detect the effect of H2S on VE-cadherin beforehand, we could not conclude that AKT itself is modified directly by H2S. Therefore, the mechanism of H2S-mediated AKT activation remains elusive.
The pulmonary endothelium serves as a semiselective barrier between the plasma and interstitium to micromolecules and bioactive agents. Endothelial injury results in barrier dysfunction, which contributes to pulmonary edema in lung injury. In this study, we used an EC permeability injury model induced by NaHS to examine the permeability of HPMECs to sodium fluoresceinand FITC-dextran. The results presented here showed that the change in EC permeability to small molecules is inversely related to the variety of claudin-5 levels, which strongly suggests that the attenuation of NaHS-induced EC permeability to small molecules by Dex is mediated by claudin-5. It appears likely that claudin-5 may play a key role in size-selective lung vascular permeability in ALI, as previously reported (Chen et al., 2014; Nitta et al., 2003).
There are some limitations in our study. Although pretreatment with Dex might exert a protective effect on H2S-mediated ALI, posttreatment may be more practical than the former treatment in clinical situations. Therefore, further studies designed to investigate the overall mechanism of the protective effect of Dex are warranted. Moreover, while this study has shown the deleterious effect of NaHS on ECs, there is a considerable need for in vivo studies.
In conclusions, collectively, the present work demonstrates that claudin-5 might be involved in the mechanism of H2S-induced ALI and Dex-mediated protective effects. The regulation of claudin-5 is partially attributed to the activation of the PI3K/AKT/FoxO1 pathway. Therefore, claudin-5 might be a potential target for the treatment of H2S-induced ALI.
Conflict of interest
The authors declare that there is no conflict of interest.
REFERENCES
- Aslam, M., Ahmad, N., Srivastava, R. and Hemmer, B. (2012): TNF-alpha induced NFκB signaling and p65 (RelA) overexpression repress Cldn5 promoter in mouse brain endothelial cells. Cytokine, 57, 269-275.
- Chen, W., Sharma, R., Rizzo, A.N., Siegler, J.H., Garcia, J.G. and Jacobson, J.R. (2014): Role of claudin-5 in the attenuation of murine acute lung injury by simvastatin. Am. J. Respir. Cell Mol. Biol., 50, 328-336.
- Crossland, H., Constantin-Teodosiu, D., Greenhaff, P.L. and Gardiner, S.M. (2010): Low-dose dexamethasone prevents endotoxaemia-induced muscle protein loss and impairment of carbohydrate oxidation in rat skeletal muscle. J. Physiol., 588, 1333-1347.
- Dalmasso, A.P., Goldish, D., Benson, B.A., Tsai, A.K., Wasiluk, K.R. and Vercellotti, G.M. (2014): Interleukin-4 induces up-regulation of endothelial cell claudin-5 through activation of FoxO1: role in protection from complement-mediated injury. J. Biol. Chem., 289, 838-847.
- Eghbal, M.A., Pennefather, P.S. and O’Brien, P.J. (2004): H2S cytotoxicity mechanism involves reactive oxygen species formation and mitochondrial depolarisation. Toxicology, 203, 69-76.
- Gross, C.M., Aggarwal, S., Kumar, S., Tian, J., Kasa, A., Bogatcheva, N., Datar, S.A., Verin, A.D., Fineman, J.R. and Black, S.M. (2014): Sox18 preserves the pulmonary endothelial barrier under conditions of increased shear stress. J. Cell. Physiol., 229, 1802-1816.
- Jang, A.S., Concel, V.J., Bein, K., Brant, K.A., Liu, S., Pope-Varsalona, H., Dopico, R.A. Jr., Di, Y.P., Knoell, D.L., Barchowsky, A. and Leikauf, G.D. (2011): Endothelial dysfunction and claudin 5 regulation during acrolein-induced lung injury. Am. J. Respir. Cell Mol. Biol., 44, 483-490.
- Jiang, L., Wang, J., Su, C., Qian, W., Chen, J., Zhu, B., Zhang, H., Xiao, H. and Zhang, J. (2014): α-ENaC, a therapeutic target of dexamethasone on hydrogen sulfide induced acute pulmonary edema. Environ. Toxicol. Pharmacol., 38, 616-624.
- Jiang, L., Wang, Y., Su, C., Sun, H., Zhang, H., Zhu, B., Zhang, H., Xiao, H., Wang, J. and Zhang, J. (2015): Epithelial sodium channel is involved in H2S-induced acute pulmonary edema. Inhal. Toxicol., 27, 613-620.
- Jin, W., Rong, L., Liu, Y., Song, Y., Li, Y. and Pan, J. (2013): Increased claudin-3, -4 and -18 levels in bronchoalveolar lavage fluid reflect severity of acute lung injury. Respirology, 18, 643-651.
- Kage, H., Flodby, P., Gao, D., Kim, Y.H., Marconett, C.N., DeMaio, L., Kim, K.J., Crandall, E.D. and Borok, Z. (2014): Claudin 4 knockout mice: normal physiological phenotype with increased susceptibility to lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol., 307, L524-L536.
- Koval, M. (2013): Claudin heterogeneity and control of lung tight junctions. Annu. Rev. Physiol., 75, 551-567.
- Li, G., Flodby, P., Luo, J., Kage, H., Sipos, A., Gao, D., Ji, Y., Beard, L.L., Marconett, C.N., DeMaio, L., Kim, Y.H., Kim, K.J., Laird-Offringa, I.A., Minoo, P., Liebler, J.M., Zhou, B., Crandall, E.D. and Borok, Z. (2014): Knockout mice reveal key roles for claudin 18 in alveolar barrier properties and fluid homeostasis. Am. J. Respir. Cell Mol. Biol., 51, 210-222.
- Li, Q., Zhang, Q., Wang, C., Liu, X., Qu, L., Gu, L., Li, N. and Li, J. (2009): Altered distribution of tight junction proteins after intestinal ischaemia/reperfusion injury in rats. J. Cell. Mol. Med., 13 (9B), 4061-4076.
- Mehta, D., Ravindran, K. and Kuebler, W.M. (2014): Novel regulators of endothelial barrier function. Am. J. Physiol. Lung Cell. Mol. Physiol., 307, L924-L935.
- Millar, F.R., Summers, C., Griffiths, M.J., Toshner, M.R. and Proudfoot, A.G. (2016): The pulmonary endothelium in acute respiratory distress syndrome: insights and therapeutic opportunities. Thorax, 71, 462-473.
- Mitchell, L.A., Ward, C., Kwon, M., Mitchell, P.O., Quintero, D.A., Nusrat, A., Parkos, C.A. and Koval, M. (2015): Junctional adhesion molecule A promotes epithelial tight junction assembly to augment lung barrier function. Am. J. Pathol., 185, 372-386.
- Nitta, T., Hata, M., Gotoh, S., Seo, Y., Sasaki, H., Hashimoto, N., Furuse, M. and Tsukita, S. (2003): Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol., 161, 653-660.
- Nomura, K., Obata, K., Keira, T., Miyata, R., Hirakawa, S., Takano, K., Kohno, T., Sawada, N., Himi, T. and Kojima, T. (2014): Pseudomonas aeruginosa elastase causes transient disruption of tight junctions and downregulation of PAR-2 in human nasal epithelial cells. Respir. Res., 15, 21.
- Prota, L.F., Cebotaru, L., Cheng, J., Wright, J., Vij, N., Morales, M.M. and Guggino, W.B. (2012): Dexamethasone regulates CFTR expression in Calu-3 cells with the involvement of chaperones HSP70 and HSP90. PLoS One, 7, e47405.
- Suzuki, H., Tani, K., Tamura, A., Tsukita, S. and Fujiyoshi, Y. (2015): Model for the architecture of claudin-based paracellular ion channels through tight junctions. J. Mol. Biol., 427, 291-297.
- Truong, D.H., Eghbal, M.A., Hindmarsh, W., Roth, S.H. and O’Brien, P.J. (2006): Molecular mechanisms of hydrogen sulfide toxicity. Drug Metab. Rev., 38, 733-744.
- Wang, J., Zhang, H., Su, C., Chen, J., Zhu, B., Zhang, H., Xiao, H. and Zhang, J. (2014): Dexamethasone ameliorates H2S-induced acute lung injury by alleviating matrix metalloproteinase-2 and -9 expression. PLoS One, 9, e94701.
- Ward, C., Schlingmann, B., Stecenko, A.A., Guidot, D.M. and Koval, M. (2015): NF-κB inhibitors impair lung epithelial tight junctions in the absence of inflammation. Tissue Barriers, 3, e982424.
- Weksler, B.B., Subileau, E.A., Perrière, N., Charneau, P., Holloway, K., Leveque, M., Tricoire-Leignel, H., Nicotra, A., Bourdoulous, S., Turowski, P., Male, D.K., Roux, F., Greenwood, J., Romero, I.A. and Couraud, P.O. (2005): Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J., 19, 1872-1874.
- Whitsett, J.A. and Alenghat, T. (2015): Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat. Immunol., 16, 27-35.
- Wilmes, A., Aschauer, L., Limonciel, A., Pfaller, W. and Jennings, P. (2014): Evidence for a role of claudin 2 as a proximal tubular stress responsive paracellular water channel. Toxicol. Appl. Pharmacol., 279, 163-172.
- Xia, X.M., Wang, F.Y., Wang, Z.K., Wan, H.J., Xu, W.A. and Lu, H. (2010): Emodin enhances alveolar epithelial barrier function in rats with experimental acute pancreatitis. World J. Gastroenterol., 16, 2994-3001.
- Xu, C., Jiang, L., Zou, Y., Xing, J., Sun, H., Zhu, B., Zhang, H., Wang, J. and Zhang, J. (2017): Involvement of water channel Aquaporin 5 in H2S-induced pulmonary edema. Environ. Toxicol. Pharmacol., 49, 202-211.
- Zhang, Y., Guan, L., Yu, J., Zhao, Z., Mao, L., Li, S. and Zhao, J. (2016): Pulmonary endothelial activation caused by extracellular histones contributes to neutrophil activation in acute respiratory distress syndrome. Respir. Res., 17, 155.
- Zinter, M.S., Delucchi, K.L., Kong, M.Y., Orwoll, B.E., Spicer, A.S., Lim, M.J., Alkhouli, M.F., Ratiu, A.E., McKenzie, A.V., McQuillen, P.S., Dvorak, C.C., Calfee, C.S., Matthay, M.A. and Sapru, A. (2019): Early Plasma Matrix Metalloproteinase Profiles: A Novel Pathway in Pediatric Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med., 199, 181-189.