Abstract
The aryl hydrocarbon receptor (AhR) regulates expression of genes encoding drug/xenobiotic metabolizing enzymes. Cytochrome P450 (CYP) 3A5 is involved in drug metabolism. However, regulation of CYP3A5 gene expression is not yet well understood. In this study, we aimed to investigate the effect of the ligands of AhR on CYP3A5 gene expression. CYP3A5 mRNA expression was induced by the polycyclic aromatic hydrocarbons (PAHs) such as 3-methylcholanthrene (3MC) and benzo[a]pyrene in HepG2 cells. To determine whether the PAHs induced CYP3A5 gene expression via AhR, we generated AhR knockout (AhR KO) HepG2 cells. CYP3A5 mRNA expression was not induced by 3MC treatment in AhR KO cells. In addition, we generated AhR rescue cells from AhR KO cells and evaluated CYP3A5 mRNA expression. We found that CYP3A5 mRNA expression was induced by 3MC treatment in AhR rescue cells. Taken together, these results demonstrated that CYP3A5 mRNA expression was induced by PAHs via AhR in HepG2 cells. Our findings suggest that ligand-activated AhR affects CYP3A5-mediated drug metabolism.
INTRODUCTION
The aryl hydrocarbon receptor (AhR) belongs to the basic helix-loop-helix/Per-Arnt-Sim (bHLH/PAS) family and functions as a ligand-activated transcription factor (Kawajiri and Fujii-Kuriyama, 2017). Following ligand binding, AhR translocates to the nucleus and heterodimerizes with aryl hydrocarbon receptor nuclear translocator (Arnt). The AhR/Arnt heterodimer binds to specific DNA sequences called xenobiotic response elements (XREs) and up-regulates the expression of AhR target genes such as cytochrome P450 (CYP) 1A1, 1A2, and 1B1. AhR has been mainly studied in the context of its ability to mediate the toxic effects resulting from exposure to dioxins and polycyclic aromatic hydrocarbons (PAHs) (Shimizu et al., 2000; Mimura et al., 1997). Recent studies have revealed that AhR plays pivotal roles such as anti-cancer effect (Zhao et al., 2012), energy metabolism (Wada et al., 2016), immunity (Kimura et al., 2008), and drug metabolism (Aleksunes and Klaassen, 2012).
The CYP3A subfamily is comprised of four genes, CYP3A4, CYP3A5, CYP3A7, and CYP3A43, and plays a major role in the metabolism of approximately 30% of clinically used drugs (Zanger and Schwab, 2013). Similarly, the other members of the CYP3A family, CYP3A5 is expressed in the human liver. CYP3A5 gene has been found to have several single-nucleotide polymorphisms such as CYP3A5*3, CYP3A5*6 and CYP3A5*7. The CYP3A5*3 is the major allele in CYP3A5. This allele is caused due to the insertion of an early stop codon which produces a nonfunctional protein (Kuehl et al., 2001). CYP3A5*3 is observed in Europeans, Africans, East Asians, South Asians, and admixed Americans, with a frequency of 94%, 18%, 71%, 67%, and 80%, respectively (Zhou et al., 2017). The substrate specificities of CYP3A5 is overlapped with CYP3A4, but some differences in catalytic properties have been reported (Niwa et al., 2008). It has been reported that various drugs, such as atazanavir, vincristine, midazolam, vardenafil, verapamil, and tacrolimus, are mainly metabolized by CYP3A5 (Tseng et al., 2014).
It has been reported that CYP3A5 gene expression is regulated by nuclear receptors such as the glucocorticoid receptor (GR), constitutively activated receptor (CAR), and pregnane X receptor (PXR) (Hukkanen et al., 2003; Burk et al., 2004). CYP3A5 expressors have a high ability to metabolize tacrolimus and require 1.5- to 2-fold higher dosage of tacrolimus compared to the usual dosage (Birdwell et al., 2015). Clarification of the CYP3A5 expression mechanism is pivotal for personalized medicine. Since AhR plays an important role in drug metabolism, we predicted that AhR regulates CYP3A5 gene expression. We searched for XRE sequence upstream of CYP3A5 gene using a web-based tool, The University of California, Santa Cruz Genome Browser (http://www.genome.ucsc.edu/). The XRE sequence was present approximately -2.1 kb upstream from the transcription start site of the CYP3A5 gene in the human genome. In this study, we investigated the effect of AhR ligands on CYP3A5 gene expression in HepG2 cells.
MATERIALS AND METHODS
Chemicals
3-methylcholanthrene (3MC) and benzo[a]pyrene (B[a]P) were purchased from FUJIFILM Wako (Osaka, Japan).
Cell culture, generation of stable AhR knockout (KO) cells, and AhR rescue cells
HepG2 hepatocellular carcinoma cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in Dulbecco’s modified Eagle medium (Nacalai Tesque, Kyoto, Japan), supplemented with 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO, USA), 100 units/mL penicillin, and 100 µg/mL of streptomycin (Nacalai Tesque). The cells were cultured at 37°C with 5% CO2 with saturated humidity. AhR knockout (AhR KO) HepG2 cells were established according to our previously described method (Yamashita et al., 2018, 2019) with modifications. The gRNAs sequences for the human AhR gene were 5′-ccggGCGACTGGCGTAGGTGATGT-3′/5′-aaacACATCACCTACGCCAGTCGC-3′. The gRNAs were annealed and cloned into a pGuide-it vector (Clontech Laboratories, Inc., Mountain View, CA, USA). AhR rescue cells, which stably expressed myc-tagged fusion AhR, were generated by transfecting AhR KO cells with pEBMulti-Hyg/myc-AhR plasmid and subjecting the cells to serial hygromycin selections. The pEBMulti-Hyg/myc-AhR plasmid was constructed from pcDNA5/TO myc AhR plasmid, using the Infusion cloning kit (TaKaRa, Shiga, Japan), by inserting the myc tag and AhR gene into Hind III, BamH I, and EcoR V, Xho I sites, respectively.
Real-time RT-PCR
Total RNA was isolated from the cells using ISOGEN II (Nippon Gene Co. Ltd, Tokyo, Japan) according to the manufacturer’s protocol. cDNA was synthesized using the ExScript RT reagent kit (TaKaRa). Quantitative real-time RT-PCR was carried out using SYBR premix ExTaq (TaKaRa) on a Smart Cycler II system (Cepheid Inc., Sunnyvale, CA, USA). The following primer pairs were used: CYP3A5: forward 5’-GCTCGCAGCCCAGTCAATA-3’, reverse 5’-AGGTGGTGCCTTATTGGGC-3’, CYP1A1: forward 5’-GTCATCTGTGCCATTTGCTTTG-3’, reverse 5’-CAACCACCTCCCCGAAATTATT-3’ and ribosomal protein lateral stalk subunit P0 (RPLP0): forward 5’-GGCGACCTGGAAGTCCAACT-3’, reverse 5’-CCATCAGCACCACAGCCTTC-3’. RPLP0 was used as a control.
Western blot analysis
The cells were harvested and lysed in sodium dodecyl sulfate (SDS) sample buffer containing 125 mM Tris-HCI pH 6.8, 4% SDS, 20% glycerol, 10% dithiothreitol, and 0.01% bromophenol blue. Whole-cell lysates were resolved by SDS-polyacrylamide gel electrophoresis and subsequently transferred onto a polyvinylidene fluoride membrane (Immobilon; Millipore, Burlington, MA, USA). Immunoblotting was performed using anti-AhR (D5S6H) (1:2000 dilution; Cell Signaling Technology, Danvers, MA, USA) and anti-β actin (1:500 dilution; Santa Cruz Biotechnology, Dallas, TX, USA) as the primary antibodies. Horseradish peroxidase-conjugated anti-rabbit and anti-mouse IgG (1:4000 dilution; Cell Signaling Technology) were used as secondary antibodies. Band intensities were detected using a LumiVision PRO 400EX (Aisin Seiki, Aichi, Japan) and analyzed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Plasmid construction
pGL3-XRE, an XRE-driven firefly luciferase-expressing plasmid, was previously constructed by inserting three tandem repeats of the consensus XRE-containing sequence (5’-TCTTCTCACGCAACTCCGA-3’) into the upstream region of the firefly luciferase gene in the pGL3-promoter vector (Promega, Madison, WI, USA) (Sanada et al., 2009). The underlined nucleotides are the consensus XRE.
Luciferase reporter analysis
Using PEI Max reagent (Polysciences, Warrington, PA, USA), cells were co-transfected with both pGL3-XRE luciferase reporter plasmid and pGL4.74 (hRluc/TK; Promega, San Luis Obispo, CA, USA) carrying the Renilla luciferase gene as an internal standard. Following overnight culture in growth medium, the cells were treated with 3MC for 24 hr. The cells were then lysed with passive lysis buffer and luciferase activity was measured using the Dual Luciferase Reporter Assay System (Promega) according to the manufacturer’s protocol. The activities of firefly luciferase were normalized to those of Renilla luciferase.
Statistical analysis
Statistical comparisons were performed using one-way analysis of variance followed by Dunnett’s multiple comparison test as the post-hoc test. Unpaired Student’s t-test was performed to compare the differences between two groups. Results with p < 0.05 were considered as statistically significant. Statistical analyses performed using KaleidaGraph software, version 4.1.1 (Synergy Software, Eden Prairie, MN, USA).
RESULTS AND DISCUSSION
In silico analysis revealed the XRE sequence was present approximately -2.1 kb upstream from the transcription start site of the CYP3A5 gene in the human genome. To determine the effects of AhR ligands, 3MC and B[a]P, on CYP3A5 gene expression, we measured the mRNA expression levels of CYP3A5 and the typical AhR target gene, CYP1A1, in HepG2 cells. Both CYP3A5 and CYP1A1 mRNA expression levels were increased by 3MC (1 µM) or B[a]P (2 µM) treatment (Fig. 1A), and the effect of 3MC was seen in a dose-dependent manner (Fig. 1B). In general, AhR binds to XRE in the promoter region of the gene thereby enhancing its transcription. Therefore, we constructed a reporter luciferase plasmid with the CYP3A5 gene promoter region (-2463/-1 bp) containing XRE and evaluated the effect of 3MC on luciferase activity. The luciferase activity was not elevated by 3MC treatment (data not shown). This result suggests that AhR regulates CYP3A5 gene expression via other regions containing XRE, such as intron of CYP3A5 gene or distant from CYP3A5 gene. AhR may also regulate the expression and/or activating transcription factors involved in the induction of CYP3A5 gene expression. Further investigation is needed to clarify the mechanism of induction of CYP3A5 gene expression by AhR.

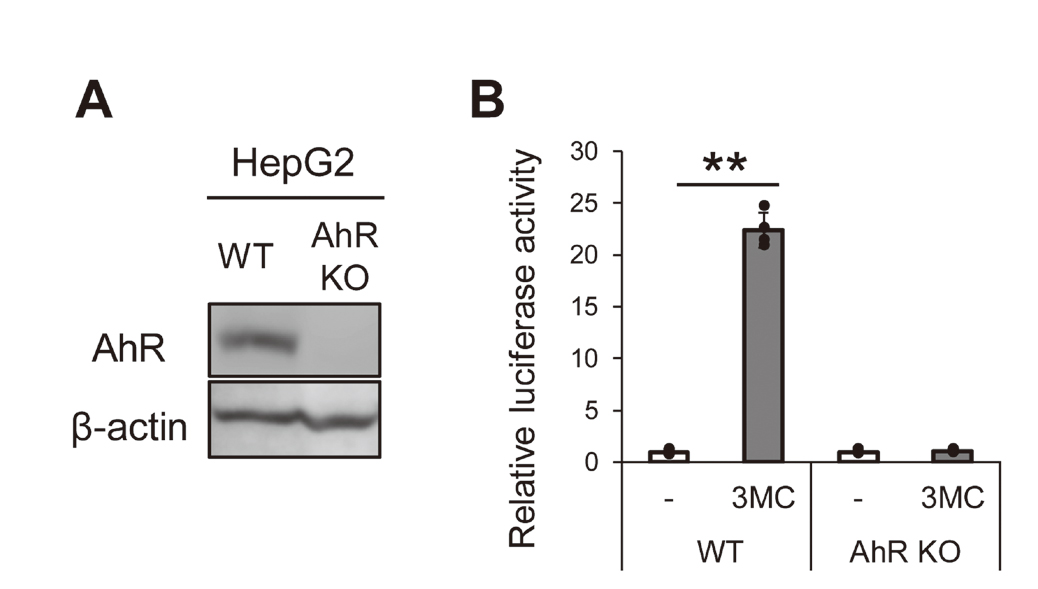
It has been reported that the induction of CYP3A4 gene expression by PAHs occurs via PXR, but not via AhR, in HepG2 cells (Kumagai et al., 2012). Therefore, we established AhR knockout (AhR KO) HepG2 cells, using the CRISPR/Cas9 system, to examine whether activated AhR was involved in the up-regulation of CYP3A5 gene expression. AhR protein was not detected in AhR KO cells as shown in Fig. 2A. Furthermore, XRE-driven luciferase activity was increased by 3MC in wild type (WT) HepG2 cells, but not in AhR KO cells (Fig. 2B). These results confirmed that AhR KO cells were established. Next, we evaluated the mRNA expression of CYP1A1 and CYP3A5 in AhR KO cells. CYP1A1 and CYP3A5 mRNA expression levels were not increased by 3MC treatment (Fig. 3).

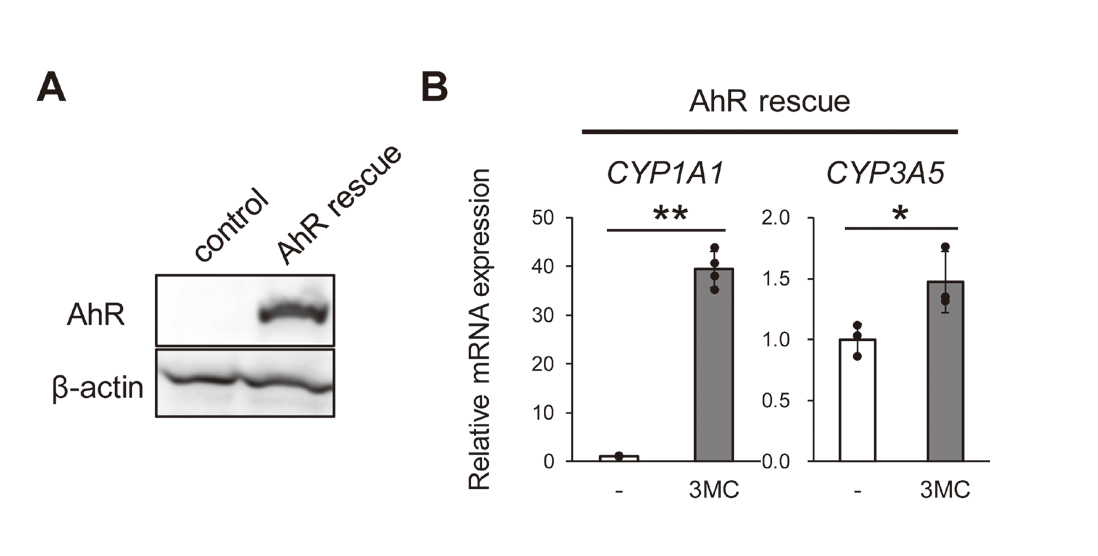
The CRISPR/Cas9 system has an off-target effect in which other genes can be possibly edited (Zhang et al., 2015). Therefore, we established two kinds of cells from AhR KO cells: AhR rescue cells and control cells. The former is derived by transfecting the AhR KO cells with an episomal vector containing the AhR gene (AhR rescue), and the latter by transfecting the cells with an empty vector (control cells). As shown in Fig. 4A, AhR protein expression was observed in AhR rescue cells, but not in control cells. In addition, the mRNA expression levels of CYP1A1 and CYP3A5 were significantly increased by 3MC in AhR rescue cells (Fig. 4B). These results indicated that the results shown in Fig. 3 were not due to the off-target effects of the CRISPR/Cas9 system, and that induction of CYP3A5 mRNA expression requires AhR expression.
PAHs are ubiquitous environmental pollutants produced during the incomplete combustion of organic materials. It is also known that cigarette smoke contains various PAHs and activates AhR. Based on our results, we predicted that smokers might have elevated CYP3A5 gene expression compared to non-smoker. However, one study suggested that cigarette smoking decreased the mRNA levels of CYP3A5 in alveolar macrophages (Hukkanen et al., 2003). Therefore, further studies are needed to determine the mechanism of regulation of CYP3A5 gene expression by AhR in various cell types or tissues.
Taken together, the present study demonstrated that PAHs induced CYP3A5 mRNA expression in HepG2 cells via AhR. Further studies on the molecular mechanism of CYP3A5 up-regulation by AhR may provide useful information for personalized medicine.
ACKNOWLEDGMENTS
This work was supported by JSPS KAKENHI Grant Numbers 19K23811 (to N.Y.).
Conflict of interest
The authors declare that there is no conflict of interest.
REFERENCES
- Aleksunes, L.M. and Klaassen, C.D. (2012): Coordinated regulation of hepatic phase I and II drug-metabolizing genes and transporters using AhR-, CAR-, PXR-, PPARα-, and Nrf2-null mice. Drug Metab. Dispos., 40, 1366-1379.
- Birdwell, K.A., Decker, B., Barbarino, J.M., Peterson, J.F., Stein, C.M., Sadee, W., Wang, D., Vinks, A.A., He, Y., Swen, J.J., Leeder, J.S., van Schaik, R., Thummel, K.E., Klein, T.E., Caudle, K.E. and MacPhee, I.A. (2015): Clinical Pharmacogenetics Implementation Consortium (CPIC) Guidelines for CYP3A5 Genotype and Tacrolimus Dosing. Clin. Pharmacol. Ther., 98, 19-24.
- Burk, O., Koch, I., Raucy, J., Hustert, E., Eichelbaum, M., Brockmöller, J., Zanger, U.M. and Wojnowski, L. (2004): The induction of cytochrome P450 3A5 (CYP3A5) in the human liver and intestine is mediated by the xenobiotic sensors pregnane X receptor (PXR) and constitutively activated receptor (CAR). J. Biol. Chem., 279, 38379-38385.
- Hukkanen, J., Väisänen, T., Lassila, A., Piipari, R., Anttila, S., Pelkonen, O., Raunio, H. and Hakkola, J. (2003): Regulation of CYP3A5 by glucocorticoids and cigarette smoke in human lung-derived cells. J. Pharmacol. Exp. Ther., 304, 745-752.
- Kawajiri, K. and Fujii-Kuriyama, Y. (2017): The aryl hydrocarbon receptor: a multifunctional chemical sensor for host defense and homeostatic maintenance. Exp. Anim., 66, 75-89.
- Kimura, A., Naka, T., Nohara, K., Fujii-Kuriyama, Y. and Kishimoto, T. (2008): Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc. Natl. Acad. Sci. USA, 105, 9721-9726.
- Kuehl, P., Zhang, J., Lin, Y., Lamba, J., Assem, M., Schuetz, J., Watkins, P.B., Daly, A., Wrighton, S.A., Hall, S.D., Maurel, P., Relling, M., Brimer, C., Yasuda, K., Venkataramanan, R., Strom, S., Thummel, K., Boguski, M.S. and Schuetz, E. (2001): Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat. Genet., 27, 383-391.
- Kumagai, T., Suzuki, H., Sasaki, T., Sakaguchi, S., Miyairi, S., Yamazoe, Y. and Nagata, K. (2012): Polycyclic aromatic hydrocarbons activate CYP3A4 gene transcription through human pregnane X receptor. Drug Metab. Pharmacokinet., 27, 200-206.
- Mimura, J., Yamashita, K., Nakamura, K., Morita, M., Takagi, T.N., Nakao, K., Ema, M., Sogawa, K., Yasuda, M., Katsuki, M. and Fujii-Kuriyama, Y. (1997): Loss of teratogenic response to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice lacking the Ah (dioxin) receptor. Genes Cells, 2, 645-654.
- Niwa, T., Murayama, N., Emoto, C. and Yamazaki, H. (2008): Comparison of kinetic parameters for drug oxidation rates and substrate inhibition potential mediated by cytochrome P450 3A4 and 3A5. Curr. Drug Metab., 9, 20-33.
- Sanada, N., Gotoh, Y., Shimazawa, R., Klinge, C.M. and Kizu, R. (2009): Repression of activated aryl hydrocarbon receptor-induced transcriptional activation by 5α-dihydrotestosterone in human prostate cancer LNCaP and human breast cancer T47D cells. J. Pharmacol. Sci., 109, 380-387.
- Shimizu, Y., Nakatsuru, Y., Ichinose, M., Takahashi, Y., Kume, H., Mimura, J., Fujii-Kuriyama, Y. and Ishikawa, T. (2000): Benzo[a]pyrene carcinogenicity is lost in mice lacking the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA, 97, 779-782.
- Tseng, E., Walsky, R.L., Luzietti, R.A. Jr., Harris, J.J., Kosa, R.E., Goosen, T.C., Zientek, M.A. and Obach, R.S. (2014): Relative contributions of cytochrome CYP3A4 versus CYP3A5 for CYP3A-cleared drugs assessed in vitro using a CYP3A4-selective inactivator (CYP3cide). Drug Metab. Dispos., 42, 1163-1173.
- Wada, T., Sunaga, H., Miyata, K., Shirasaki, H., Uchiyama, Y. and Shimba, S. (2016): Aryl Hydrocarbon Receptor Plays Protective Roles against High Fat Diet (HFD)-induced Hepatic Steatosis and the Subsequent Lipotoxicity via Direct Transcriptional Regulation of Socs3 Gene Expression. J. Biol. Chem., 291, 7004-7016.
- Yamashita, N., Kanno, Y., Saito, N., Terai, K., Sanada, N., Kizu, R., Hiruta, N., Park, Y., Bujo, H. and Nemoto, K. (2019): Aryl hydrocarbon receptor counteracts pharmacological efficacy of doxorubicin via enhanced AKR1C3 expression in triple negative breast cancer cells. Biochem. Biophys. Res. Commun., 516, 693-698.
- Yamashita, N., Saito, N., Zhao, S., Terai, K., Hiruta, N., Park, Y., Bujo, H., Nemoto, K. and Kanno, Y. (2018): Heregulin-induced cell migration is promoted by aryl hydrocarbon receptor in HER2-overexpressing breast cancer cells. Exp. Cell Res., 366, 34-40.
- Zanger, U.M. and Schwab, M. (2013): Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther., 138, 103-141.
- Zhang, X.-H., Tee, L.Y., Wang, X.-G., Huang, Q.-S. and Yang, S.-H. (2015): Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol. Ther. Nucleic Acids, 4, e264.
- Zhao, S., Kanno, Y., Nakayama, M., Makimura, M., Ohara, S. and Inouye, Y. (2012): Activation of the aryl hydrocarbon receptor represses mammosphere formation in MCF-7 cells. Cancer Lett., 317, 192-198.
- Zhou, Y., Ingelman-Sundberg, M. and Lauschke, V.M. (2017): Worldwide Distribution of Cytochrome P450 Alleles: A Meta-analysis of Population-scale Sequencing Projects. Clin. Pharmacol. Ther., 102, 688-700.