Abstract
Excessive use of Ketamine (KET) has a neurotoxic effect on the brain. This study explored the effect of Transient Receptor Potential Vanilloid 4 (TRPV4) on KET-induced neurotoxicity in the hippocampus. We extracted and identified rat hippocampal neuronal cells. The hippocampal neurons were treated with different concentrations (0, 0.1, 1, 10, 100, 300 and 1000 μmol/L) of KET (6, 12 and 24 hr). Cell viability was detected by cell counting Kit-8 (CCK-8), and TRPV4 expression was detected by quantitative Real Time-Polymerase Chain Reaction (qRT-PCR) and western blot. After silencing TRPV4, we tested cell viability and apoptosis. The contents of superoxide dismutase (SOD), glutathione (GSH), malondialdehyde (MDA), and catalase (CAT) were detected by colorimetry, and the contents of TNF-α, IL-1β, IL-6 and reactive oxygen species (ROS) were detected by Enzyme-Linked ImmunoSorbent Assay (ELISA). Finally, the expression levels of apoptosis-related proteins Bcl-2, Bax and Cleaved caspase-3, and phosphorylated-p65 (p-65), p65, phosphorylated-IκBα (p-IκBα) and IκBα were detected by qRT-PCR and western blot. KET inhibited the viability of hippocampal neurons in a dose-dependent manner, and up-regulated TRPV4 expression. SiTRPV4 inhibits KET-induced decrease in cell viability and promotes apoptosis. SiTRPV4 reduced MDA and ROS content, increased SOD, GSH and CAT levels. The release of proinflammatory factors TNF-α, IL-1β and IL-6 was also inhibited by siTRPV4. In addition, siTRPV4 up-regulated KET-induced Bcl-2 expression in hippocampal neurons, down-regulated Bax and Cleaved caspase-3, and inhibited the activation of the inflammatory pathway. Silencing TRPV4 partially reverses the neurotoxic effects induced by KET through regulating apoptosis-related proteins and p65/IκBα pathway.
INTRODUCTION
Ketamine (KET) is a derivative of N-1-phenycyclohexy-piperidine (PCP) and is a non-competitive antagonist of the N-methyl-D-aspartate (NMDA) receptor (Gao et al., 2016). KET can inhibit the thalamus-neocortical system, selectively block pain and exert analgesic effect (Mion and Villevieille, 2013). In view of the significant analgesic effect of KET, in 1964 in the United States, KET replaced PCP as a general anesthetic commonly used for surgical anesthesia (Domino et al., 1965). Due to its advantages of rapid induction and recovery, hemodynamic stability, etc., it is a widely used pediatric anesthetic (Mizrak et al., 2010; Neuhäuser et al., 2010). However, a large number of studies in recent years have shown that KET has obvious toxic side effects on the nervous system, cardiovascular system and urinary system (Morgan and Curran, 2012). Scientists have found that a small amount of KET can cause a large number of deaths in neurons in the brain of newborn rats, a change in apoptosis, and a serious cognitive, learning and memory retention disorder in adulthood (Fredriksson et al., 2004). Whether KET affects the intellectual development of infants and young children has become a constant concern of parents.
The hippocampus is a special structure formed by the central nervous system (Knierim, 2015). The hippocampal formation is not only involved in neurodegenerative diseases such as Alzheimer’s disease, dementia, but is also the area’s most sensitive to hypoxia in brain tissue (Bartsch and Wulff, 2015). Therefore, the extraction of hippocampal neuronal cells is a necessary condition for the study of neuron-related diseases (Yassa and Stark, 2011). Research shows that an acute low dose of ketamine can decrease neural plasticity in the hippocampus-to-prefrontal cortex pathway (Rame et al., 2017). Ketamine treatment can induce pyroptosis in mouse primary hippocampal neurons, and is toxic to hippocampal neurons (Ye et al., 2018).
Transient receptor potential (TRP) channels are a class of channel proteins widely expressed in cells and tissues. They are an important class of non-selective cation channels located on the cell membrane (Venkatachalam and Montell, 2007). The TRP family can be divided into seven subfamilies of Melastatin (TRPM), Canonical (TRPC), Vanilloid (TRPV), Mucolipin (TRPML), Polycystin (TRPP), NOMP-C (TRPN) and Ankyrin (TRPA) (Li, 2017). Transient receptor potential vanilloid 4 (TRPV4) is a member of the TRPV channel family. Activation of the TRPV4 receptor can cause Ca2+ influx and increase intracellular free Ca2+ concentration (Grace et al., 2017). TRPV4 receptors are widely distributed in the central nervous system such as hippocampus, cerebral cortex and thalamus. Furthermore, excessive activation of TRPV4 has been detected in acute respiratory distress syndrome, cerebral ischemia and Alzheimer’s disease (Zhang et al., 2013; Morty and Kuebler 2014; Han et al., 2018). In addition, studies have shown that excessive activation of TRPV4 can increase the production and release of pro-inflammatory cytokines and reactive oxygen species, promoting inflammatory response and oxidative stress, which may cause nerve cell damage (Hong et al., 2016). Based these findings, we proposed a reasonable hypothesis: lowering the concentration of TRPV4 can inhibit the inflammatory reaction and oxidative stress, thereby reducing the hippocampal neuronal cell damage caused by KET. In this study, hippocampal neurons in neonatal rat brain tissue were extracted, the effect and mechanism of TRPV4 on KET-induced cells were explored.
MATERIALS AND METHODS
Cell extraction
The method of hippocampal neuron cell extraction in this study refers to a number of research results at home and abroad (Kaech and Banker, 2006; Facci and Skaper, 2018). Fresh healthy SD rats within 24 hr of birth were provided by the Animal Experimental Research Center of School of Medicine, Nanchang University (SPF level, female and male are not limited). The handling of animals during the experiment was in accordance with animal ethics requirements (approval number: 20180414SPH). After thoroughly disinfecting the rats with 75% alcohol, the brain tissue was extracted and placed in Hank’s Balanced Salt Solution (HBSS, C14175500BT, Gibco, Waltham, MA, USA) treated in advance in ice bath. The bilateral hippocampus of the brain tissue was separated using a microscope, and the pia mater and blood vessels were removed. 0.125% trypsin was added to the hippocampus and shaken in a 37°C water bath for 15 min. Then, trypsin was aspirated and washed twice with HBSS, and then a cell suspension was prepared by adding DMEM medium containing 10% fetal calf serum (FBS). After filtration through a 70 μM sieve, the cell suspension had a cell density of 1 × 106/mL and was inoculated into a high sugar medium containing 10% FBS, 1% sodium pyruvate (S104174-25g, Aladdin, Shanghai, China), and 1% GlutaMax (41090036, Gibco). After adherent culture for 4 hr, the medium was replaced with maintenance medium (2% B27, 1% GlutaMax Neurobasal medium). The culture solution was changed every 3 days.
Hippocampal neuronal cell identification
We identified the extracted neuronal cells by immunofluorescence staining experiments. The extracted cells were washed twice with PBS and then fixed with 4% paraformaldehyde at 4°C for 20 min. After washing again, the cells were perforated with 0.1% Triton X-100 (4°C, 15 min, V900502-100ML, Sigma, St. Louis, MO, USA). After washing, the culture dish was blocked by adding 5% goat serum (C0265, Beyotime, Nantong, China). After 30 min, Neuronal specific enolase (NSE) primary antibody (1:200, ab220216, Abcam, Camb, UK) was added to the cells overnight (4°C). After the incubation, FITC-labeled goat anti-rabbit secondary antibody (AS-28176-1-FITC, Anaspec, Fremont, CA, USA) was used for immunostaining. After 2 hr, the nuclei of hippocampal neurons were stained with 4’,6-diamidino-2-phenylindole (DAPI, 10236276001, Roche, Basel, Switzerland) for 5 min. After thorough washing with PBS, the anti-fluorescence quenching capsule (E675011-5ml, BBI, Shanghai, China) and neutral gum were used to seal the tablets. The positive expression of NSE was obtained by fluorescence microscopy.
Cell culture and processing
The identified hippocampal neurons were cultured in Neurobasal medium (211030049, Invitrogen, Carlsbad, CA, USA) containing 2% B27 and 1% GlutaMax. It was placed in an incubator containing 95% CO2 at 37°C. To determine the neurotoxicity of different concentrations of KET, different concentrations of KET (0, 0.1, 1, 10, 100, 300, 1000 μmol/L) were added to 6-well plates supplemented with hippocampal neurons. KET was dissolved using 0.9% physiological saline. Cells were treated for 6 hr, 12 hr, and 24 hr, respectively. KET was purchased from DRE in Augsburg, Germany (C14531000).
Cell Counting Kit-8 (CCK-8) assay
The viability of the differently treated hippocampal neurons was measured by the CCK-8 assay. First, 100 μL of KET-treated hippocampal neurons were seeded into 96-well plates at a concentration of 5 × 103 cells/well. After incubation for 6, 12, and 24 hr (37°C, 5% CO2), 10 μL of CCK-8 reagent (C0037, Beyotime) was added to each well and returned to the cell culture incubator. We used a microplate reader (iMark, BIO-RAD, Hercules, CA, USA) to measure the absorbance of each group of cells after 6 hr, 12 hr, and 24 hr. The wavelength was set to 450 nm.
Cell transfection
In order to explore the effect of TRPV4 on hippocampal neuronal cells after KET treatment, we needed to silence the TRPV4 gene. Therefore, we constructed a small interfering RNA expression vector for TRPV4 and transfected into cells. The sequence encoding the shRNA (small hairpin RNA) was then initiated using the Pol III promoter. The sequences of siRNA TRPV4 (siTRPV4) were as follows: sense oligo: 5’-UCAUAUCGGCUUUCUUGUGAG-3’, antisense oligo: 5’-CACAAGAAAGCCGAUAUGAGG-3’. Next we transfected siTRPV4 into cells using the LipofectamineTM 3000 (L3000015, ThermoFisher, Waltham, MA, USA). Cells were first digested with 0.25% trypsin (P5259, Abnova, Taibei, Taiwan) for 24 hr. 30 pmol of siTRPVC4 was first mixed with 50 μL of Opti-MEM reagent (31985062, Invitrogen). 3 μL of LipofectamineTM 3000 reagent was diluted with 50 μL of Opti-MEM reagent. The two mixtures were then mixed and left for 15 min (room temperature). Finally, the above mixtures were added to the cells separately. Cell transfection efficiency was measured by qRT-PCR and western blot after 48 hr. After successful transfection, transfected cells were treated with 300 μM of KET for 12 hr.
Flow cytometry
We examined changes in apoptosis by flow cytometry. The hippocampal neurons that were tested also needed to be digested with trypsin. The digested cells were centrifuged (100 g, 4°C, 5 min) and 5 mL of PBS was added. The above steps were repeated twice. 70% ethanol was added to the finally collected cell suspension, which was then placed at 4°C overnight. After washing the cells with PBS the next day, 5 μL of Annexin V-FITC (C1062M, Beyotime) and 10 μL of propidium iodide (PI, C1062M, Beyotime) were added to the cells. The above mixture was allowed to stand in the dark for 20 min and then placed in a CytoFLEX flow cytometer (Backman Coulter, Brea, CA, USA) for detection.
Detection of oxidative stress-related factors
Colorimetry is a method of quantitative analysis. It is based on the color reaction of the colored compound to determine the content of the component to be tested by comparing or measuring the color depth of the colored substance solution. Therefore, we examined the levels of oxidative stress-related factors by colorimetry: superoxide dismutase (SOD), glutathione (GSH), malondialdehyde (MDA), catalase (CAT). The cells to be tested were first digested. The experiments were then carried out according to the instructions and reagents in the SOD (11305, AAT Bioquest, Sunnyvale, CA, USA), GSH (ml016833, Mlbio, Shanghai, China), MDA (ml022446, Mlbio), CAT (ml022379, Mlbio) test kits. Finally, the absorbance was measured by GENESYSTM 30 Visible Spectrophotometer (840-277000, ThermoFisher), and the content of different factors was calculated.
Enzyme-Linked ImmunoSorbent Assay (ELISA)
ELISA refers to a solid phase adsorption assay based on an enzyme as a label and based on an immunological binding reaction between an antigen and an antibody. We selected ELISA to analyze the levels of Tumor Necrosis Factor-α (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6) and reactive oxygen species (ROS). The cells to be tested were first subjected to a 24-hr digestion process. The cell supernatant was then extracted. According to the kit instructions, TNF-α (PT516, Beyotime), IL-1β (PI303, Beyotime), IL-6 (PI328, Beyotime) and ROS (CSB-E15037r, Cusabio, Wuhan, China) were detected in the cell supernatant. Finally, the absorbance was measured by a spectrophotometer, and the content of different factors was calculated.
Total RNA extraction and Quantitative Real Time-Polymerase Chain Reaction (qRT-PCR)
Total RNA in hippocampal neuronal cells needed to be extracted using the Trizol method. The cells were washed with PBS and then 1 mL Trizol (15596018, Invitrogen) was added. Blowing evenly with a pipette can help the cells to fully lyse. After centrifugation, chloroform (10006818, Sinopharm, Beijing, China) was added, followed by isopropanol (I112020-500 mL, Aladdin). The resulting total RNA was dissolved and the concentration was measured using a NanoDropTM 8000 spectrophotometer (ND-8000-GL, ThermoFisher). Next, the process required the use of reverse transcription k1622 RevertAid First Strand cDNA Synthesis Kit (ThermoFisher). The cDNA obtained by reverse transcription was diluted ten times with DEPC water and used. The primers (Sangon Biotech, Shanghai, China) used for qRT-PCR also needed to be diluted in advance according to the instructions. The sequences of the primers were as follows: TRPV4-F, 5’-CGTCGATGGCTCCTTCCAGT-3’, and TRPV4-R, 5’-CAGGACCAGGGCAAAGACCA-3’; Bcl-2-F, 5’-GATGACTGAGTACCTGAACC-3’, and Bcl-2-R, 5’-CTGCTTTAGTGAACCTTTTC-3’; Bax-F, 5’-GATGAACTGGACAACAACAT-3’, and Bax-R, 5’-TGCTAGCAAAGTAGAAAAGG-3’; Cleaved caspase-3-F, 5’-TGTGTGATTCTAAGTCATGG-3’, and Cleaved caspase-3-R, 5’-ATATCATCGTCAGTTCCACT-3’; β-actin-F, 5’-GTGGGTATGGGTCAGAAG-3’, and β-actin-R, 5’-GGTGTTGAAGGTCTCAAA-3’. The qRT-PCR reaction system was added in a 96-well plate: 10 μL of SYBR reagent (4913914001-1, Roche), 2 μL of primer, 2 μL of cDNA, and 6 μL of DEPC water. The following amplification conditions were set by a PCR machine (4375305, ThermoFisher): predenaturation at 95°C for 10 min, denaturation at 95°C for 15 sec, annealing at 60°C for 1 min, 40 cycles. The experimental results are represented by 2-ΔΔCT (Adnan et al., 2011). β-actin was used as an internal reference in this experiment.
Western blot
We first extracted the total protein in hippocampal neurons according to a previous study (Kim, 2017). the hippocampal neurons were washed 3 times with PBS and then 100 μL of lysate (R0020-100ml, Solarbio, Beijing, China) was added. The cells were repeatedly pipetted with a pipette to help them fully lyse. The supernatant collected after centrifugation was the protein stock we needed. The concentration of the protein stock was detected by the BCA method. Next, 100 μg of protein was separated and transferred to the nitrocellulose filter membrane (NC membrane, P-N66485-30cm*3m, Solarbio) using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The protein-transferred NC membrane was washed 3 times with 1x TBST (10 min/time), and the desired bands were cut according to the molecular weights of Marker (PR1800-10, Solarbio) and the primary antibody. The cut NC membrane was blocked in a 5% blocking solution. The above steps were all done at room temperature. After 2 hr, the blocking solution was washed off and the primary antibody was incubated with the corresponding strips (4°C). The primary antibody was recovered after 24 hr and the NC membrane was washed with 1x TBST (3 times). Next, according to the species source of the primary antibody, the corresponding secondary antibodies (Anti-Mouse, ab6728, 1:10000, Abcam; Anti-Rabbit, ab6721, 1:10000, Abcam) were added to the NC membrane. The NC membrane was incubated for 1 hr at room temperature and then washed again with 1x TBST (3 times). Finally, ECL luminescent liquid (PW0121-50ml, Leagene, Beijing, China) was added dropwise to each NC membrane. After 30 sec of reaction, the gray value of the gene of interest was detected in a GelDoc XR Biorad (Bio-rad). β-actin was used as an internal reference in this experiment. The following were the primary antibodies used: TRPV4 (1:1000, 100kD, #ab39260, Abcam), Bcl-2 (1:1000, #4223, 26kD, CST), Bax (1:1000, #ab32503, 21kD, Abcam), Cleaved caspase-3 (1:1000, #ab2302, 17kD, Abcam), Phosphorylated-p65 (p-65, 1:2000, 70 kD, #ab86299, Abcam), p65 (1:1000, 64 kD, ab16502, Abcam), Phosphorylated-IκBα (p-IκBα, 1:10000, 40kD, #ab133462, Abcam), IκBα (1:1000, 35kD, #ab32518, Abcam), β-actin (1:500, #ab8226, 42kD, Abcam).
Statistical analysis
All data from the study were analyzed using SPSS 19.0 (SPSS Inc., Chicago, IL, USA). The representation of the statistical results used the mean ± mean standard deviation. Student two-tailed t-test was used to compare differences between the two groups; one-way analysis of variance (ANOVA) followed by Tukey’s t-test was used to compare differences between more than two groups. A statistical difference was considered when p < 0.05.
RESULTS
TRPV4 is highly expressed in KET-treated hippocampal neurons
We first extracted hippocampal neuronal cells from rat brain tissue and identified them by immunofluorescence staining. NSE is an acidic protease unique to neurons and neuroendocrine cells. The labeled cells appeared green under a fluorescence microscope, while the nuclei stained with DAPI were blue. As shown in Fig. 1, the cells we extracted were hippocampal neurons. To study the toxic side effects of KET on cells, we used different concentrations (0, 0.1, 1, 10, 100, 300 and 1000 μmol/L) of KET to treat hippocampal neurons (6, 12 and 24 hr) and tested cell viability (Fig. 2A). Low concentrations of KET favored an increase in cell viability but were not significant; high concentrations of KET obviously inhibited cell viability (100, 300, 1000 μmol/L). We found that 100 μmol/L of KET had a weak inhibitory effect on cell viability, while 1000 μmol/L had a significant inhibitory effect on cell viability as early as 6 hr, and the inhibitory effect was too strong. Therefore, we chose a concentration with moderate inhibition (300 μmol/L) to continue the study. Then, we treated the cells with 300 μmol/L KET for 12 hr, and detected the expression of TRPV4 by qRT-PCR and western blot (Fig. 2B-D). The results showed that KET at 300 μmol/L significantly up-regulated the expression of TRPV4 in hippocampal neurons (Fig. 2B-D, p < 0.001).

Next, we took a measure of silencing TRPV4 to study its effect on the neurotoxic effects induced by KET (Fig. 3A-C). SiTRPV4 increased cell viability by CCK-8 assay and effectively inhibited KET-induced decrease in cell viability (Fig. 3D, p < 0.001). Flow cytometry experiments also confirmed that although KET treatment increased the apoptotic rate, siTRPV4 effectively inhibited KET-induced apoptosis (Fig. 4, p < 0.001). These findings suggest that siTRPV4 inhibits KET-induced decrease in cell viability and increase in apoptosis.
SiTRPV4 is able to inhibit KET-induced oxidative stress and proinflammatory factor release
In the detection of factors related to oxidative stress (Fig. 5A-E), we found that KET increased the ROS content and MDA, and decreased the contents of antioxidant enzymes SOD, GSH and CAT. But siTRPV4 significantly increased the antioxidant activity of cells and inhibited the oxidative stress induced by KET. We also detected the levels of proinflammatory factors TNF-α, IL-1β and IL-6 in cell culture media by ELISA. KET increased the levels of TNF-α, IL-1β and IL-6 (Fig. 5F-H, p < 0.001). However, silencing of TRPV4 obviously inhibited KET-induced production and release of pro-inflammatory factors (Fig. 5F-H, p < 0.001). This indicates that siTRPV4 is capable of inhibiting KET-induced oxidative stress and proinflammatory factor release.
SiTRPV4 can inhibit the expression of apoptotic proteins in hippocampal neurons induced by KET
Based on previous experimental results, we examined the expression of apoptosis-related genes in cells of different treatment groups (Fig. 6). Long-term exposure to KET down-regulated the expression of Bcl-2 in hippocampal neurons (Fig. 6 A-G, p < 0.001), while the expression of Bax and Cleaved caspase-3 was up-regulated (Fig. 6 A-G, p < 0.001). SiTRPV4 had the opposite regulatory effect. It up-regulated Bcl-2 and inhibited the expression of Bax and Cleaved caspase-3, effectively inhibiting KET-induced apoptosis. (Fig. 6 A-G, p < 0.001).
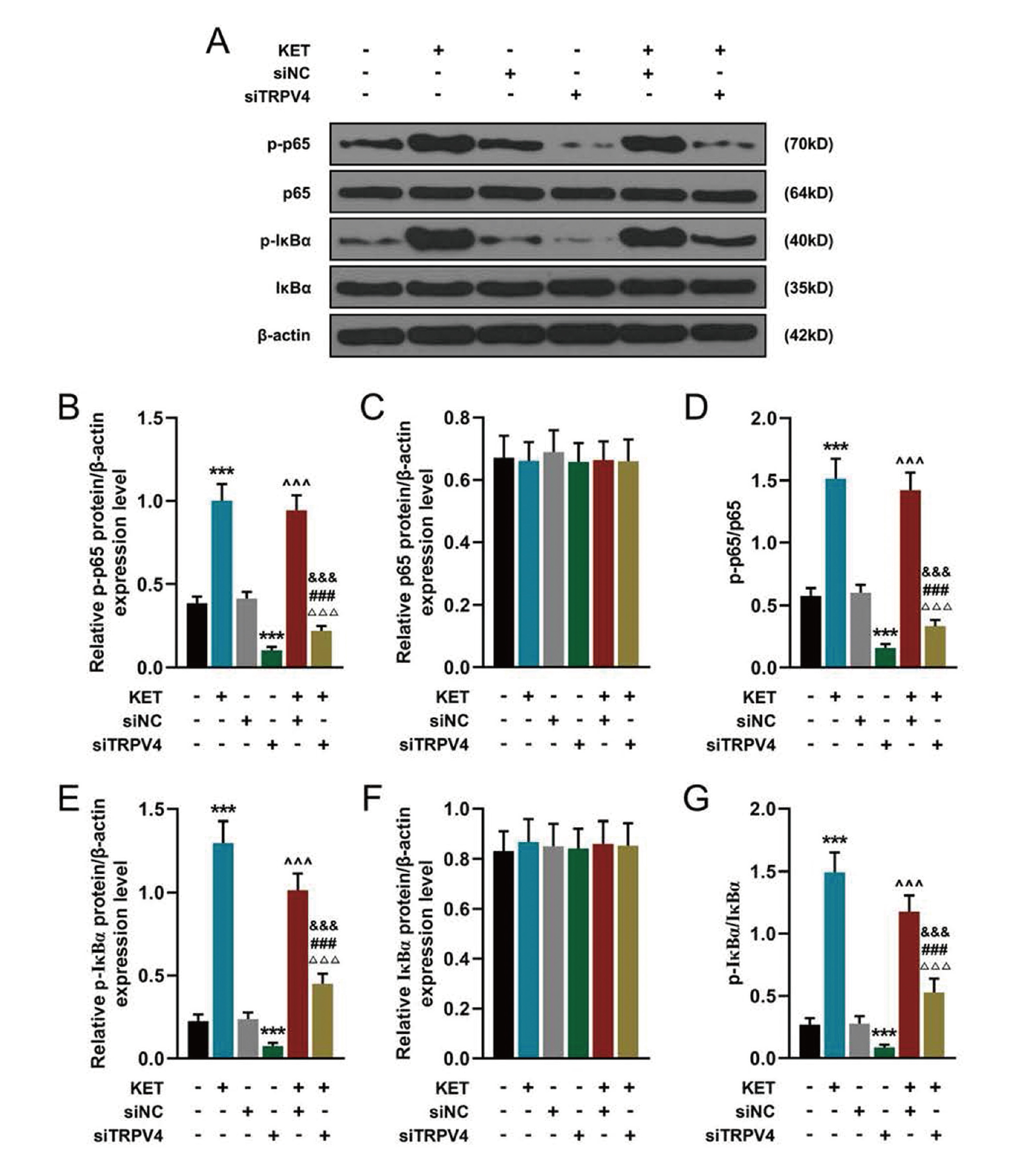
SiTRPV4 is able to inhibit KET-induced activation of the inflammatory pathway
We again analyzed the protein expression levels of p65 and IκBα in the nuclear factor kappa-B (NF-κB) inflammatory pathway by western blot. As shown in Fig. 7, in the KET-treated hippocampal neurons, both p-p65 and p-IκBα were significantly up-regulated; however, their total protein levels of p65 and IκBα did not change. Therefore, the inflammatory pathway was activated. However, this effect was obviously inhibited by siTRPV4, and the expression of p-p65 and p-IκBα was inhibited (Fig. 7 A-G, p < 0.001). It demonstrates that siTRPV4 significantly inhibits KET-induced activation of the inflammatory pathway (Fig. 7 A-G, p < 0.001).
DISCUSSION
Liu (Liu et al., 2018) has found that TRPV4 antagonists can inhibit glial cell activation and the production of TNF-α and interleukin-1β (IL-1β) and this effect is due to inhibition of phosphorylation of p65 by TRPV4-antagonists. Ohsaki indicates that keratinocytes irradiated by gamma rays induce ATP release by activating TRPV4 channels, and then ATP further activates the p38 MAPK-NF-κB pathway, thereby producing IL-6 and IL-8 (Ohsaki et al., 2018). In this research, it was found that silencing of TRPV4 promoted cell viability, reduced the levels of apoptosis, inflammatory factors and ROS.
ROS can bind directly to membrane lipids, proteins or nucleic acids, causing cell dysfunction, protein peroxidation or DNA damage through apoptosis or necrosis procedures, and causing cell damage or even death (Sinha and Dabla, 2015). MDA is the end product of oxidative stress. The determination of MDA content can reflect the degree of lipid peroxidation in tissues, which indirectly reflects the degree of oxidative damage in tissue cells. In addition, there is a class of antioxidant enzymes that play an important role in oxidative stress (Owen and Butterfield, 2010; Sies, 2015; Glorieux and Calderon, 2017): SOD can catalyze the disproportionation of superoxide anion radical (O2-) to form oxygen and hydrogen peroxide, which plays a vital role in the body’s oxidation and antioxidant balance; GSH is a tripeptide containing γ-amide bond and sulfhydryl group, which has the function of maintaining normal immune system function and anti-oxidation; CAT is an enzyme scavenger that catalyzes the decomposition of H2O2 to H2O and O2, preventing H2O2 from producing harmful -OH. Our results showed that KET-treated cells increased MDA and ROS levels, and SOD, GSH and CAT levels decreased, indicating that KET enhanced oxidative stress in hippocampal neurons. However, after treatment with siTRPV4, the anti-oxidant activity of the cells was significantly increased.
Bcl-2, Bax and Cleaved Caspase-3 are all key genes regulating apoptosis. Bcl-2 and Bax in the Bcl-2 family are extremely important in apoptosis (Hoetelmans et al., 2000). Bcl-2 encodes a proto-oncogene that is an anti-apoptotic protein; Bax is a pro-apoptotic protein of the Bcl-2 family and has a pro-apoptotic effect. In the normal physiological state of the body, the two genes are in equilibrium, but when the cells are stimulated and damaged, the equilibrium state is destroyed, causing excessive apoptosis or excessive inhibition of apoptosis. Cleaved Caspase-3 is a downstream regulatory gene of the Bcl-2 family and one of the important apoptotic proteins in the Caspase family (Choudhary et al., 2015). Our research has also achieved similar results. Silencing of TRVP4 increases Bcl-2 expression, decreases Bax and Cleaved Caspase-3, and ultimately reduces apoptosis.
NF-κB is a ubiquitous and important transcription factor in human body. It was extracted from mature B cells in 1986 and specifically binds to the enhancer κB sequence of the immunoglobulin kappa light chain gene (Hayden and Ghosh, 2011). The active form of NF-κB is a dimer, and the most common and most characteristic dimer is p50/p65 (Zhang et al., 2017). It is widely expressed in the central nervous system and plays an important role in the regulation of gene expression. In most quiescent cells, NF-κB binds to its repressor IκBα protein (Mitchell et al., 2016). This trimer composed of NF-κB and its inhibitor can cover the nuclear recognition site on NF-κB and inhibit its activity. However, when NF-κB is activated by stimuli, these stimuli can be degraded by phosphorylation of IκBα, and dissociated from NF-κB and exposed its nuclear recognition site, thereby promoting transcription of target genes (Mitchell et al., 2016). Thus phosphorylation of IκBα is a critical step in the activation of the NF-κB pathway. Studies have shown that under the induction of TNF-α, the activation of NF-κB can be promoted by accelerating the phosphorylation of p65 and IκBα, and the activated NF-κB enters the nucleus to regulate the expression of related genes (Chen et al., 2001; Green, 2003). This is consistent with our findings: in KET-induced hippocampal neuronal cells, phosphorylation of p65 and IκBα was increased. Furthermore, silencing of TRPV4 significantly reversed the induction of KET and decreased the phosphorylation of p65 and IκBα.
Studies have shown that ginsenoside Rg3 inhibits NF-κB activation, decreases the expression of NF-κB/p65 and Bcl-2, up-regulates the expression of Bax and Cleaved Caspase-3, and finally promotes the apoptosis of breast cancer cells. Additionally, an increase in ROS can further activate NF-κB and promote the release of inflammatory factors (Wei et al., 2014); IκB inhibitors and NF-κB inhibitors can inhibit lipopolysaccharide-induced cellular oxidative stress (Zuo et al., 2017); in patients with hyperglycemia, increased ROS content can activate NF-κB signaling pathway and AP-1, promote transcriptional translation of downstream gene TNF-α, and up-regulate the expression level of TNF-α (Guha et al., 2000). These findings demonstrate that NF-κB is involved in the apoptosis and oxidative stress process. It also explains that siTRPV4 may protect hippocampal neurons through inhibiting oxidative stress by decreasing the phosphorylation levels of p65 and IκBα. Nevertheless, more experiments should be performed to explore the molecular mechanism of TRPV4 regulating NF-κB pathway in hippocampal neurons.
The limitation of this study was not studying the influence of ketamine on TRPV4 channels, the restoration of TRPV4 expression in siRNA-treated cells, nor using TRPV4 antagonists, which would be conducted in future.
Taken together, our results indicate that KET promotes apoptosis and inflammatory factor release, enhances oxidative stress, and thus produces neurotoxic effects on hippocampal neurons. Silencing TRVP4 can ameliorate the neurotoxicity of KET by inhibiting the phosphorylation of p65 and IκBα and further inhibiting the activity of NF-κB. This study provides reliable experimental evidence for reducing the neurotoxicity of KET and reducing brain damage in infants and young children.
Conflict of interest
The authors declare that there is no conflict of interest.
REFERENCES
- Adnan, M., Morton, G. and Hadi, S. (2011): Analysis of rpoS and bolA gene expression under various stress-induced environments in planktonic and biofilm phase using 2(-ΔΔCT) method. Mol. Cell. Biochem., 357, 275-282.
- Bartsch, T. and Wulff, P. (2015): The hippocampus in aging and disease: from plasticity to vulnerability. Neuroscience, 309, 1-16.
- Chen, F., Castranova, V. and Shi, X. (2001): New insights into the role of nuclear factor-kappaB in cell growth regulation. Am. J. Pathol., 159, 387-397.
- Choudhary, G.S., Al-Harbi, S. and Almasan, A. (2015): Caspase-3 activation is a critical determinant of genotoxic stress-induced apoptosis. Methods Mol. Biol., 1219, 1-9.
- Domino, E.F., Chodoff, P. and Corssen, G. (1965): Pharmacologic effects of CI-581, a new dissociative anesthetic, in man. Clin. Pharmacol. Ther., 6, 279-291.
- Facci, L. and Skaper, S.D. (2018): Culture of Rodent Cortical, Hippocampal, and Striatal Neurons. Methods Mol. Biol., 1727, 39-47.
- Fredriksson, A., Archer, T., Alm, H., Gordh, T. and Eriksson, P. (2004): Neurofunctional deficits and potentiated apoptosis by neonatal NMDA antagonist administration. Behav. Brain Res., 153, 367-376.
- Gao, M., Rejaei, D. and Liu, H. (2016): Ketamine use in current clinical practice. Acta Pharmacol. Sin., 37, 865-872.
- Glorieux, C. and Calderon, P.B. (2017): Catalase, a remarkable enzyme: targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem., 398, 1095-1108.
- Grace, M.S., Bonvini, S.J., Belvisi, M.G. and McIntyre, P. (2017): Modulation of the TRPV4 ion channel as a therapeutic target for disease. Pharmacol. Ther., 177, 9-22.
- Green, D.R. (2003): Death and NF-kappaB in T cell activation: life at the edge. Mol. Cell, 11, 551-552.
- Guha, M., Bai, W., Nadler, J.L. and Natarajan, R. (2000): Molecular mechanisms of tumor necrosis factor alpha gene expression in monocytic cells via hyperglycemia-induced oxidant stress-dependent and -independent pathways. J. Biol. Chem., 275, 17728-17739.
- Han, J., Xu, H.H., Chen, X.L., Hu, H.R., Hu, K.M., Chen, Z.W. and He, G.W. (2018): Total Flavone of Rhododendron Improves Cerebral Ischemia Injury by Activating Vascular TRPV4 to Induce Endothelium-Derived Hyperpolarizing Factor-Mediated Responses. Evid. Based Complement. Alternat. Med., 2018, 8919867.
- Hayden, M.S. and Ghosh, S. (2011): NF-κB in immunobiology. Cell Res., 21, 223-244.
- Hoetelmans, R., van Slooten, H.J., Keijzer, R., Erkeland, S., van de Velde, C.J. and Dierendonck, J.H. (2000): Bcl-2 and Bax proteins are present in interphase nuclei of mammalian cells. Cell Death Differ., 7, 384-392.
- Hong, Z., Tian, Y., Yuan, Y., Qi, M., Li, Y., Du, Y., Chen, L. and Chen, L. (2016): Enhanced Oxidative Stress Is Responsible for TRPV4-Induced Neurotoxicity. Front. Cell. Neurosci., 10, 232.
- Kaech, S. and Banker, G. (2006): Culturing hippocampal neurons. Nat. Protoc., 1, 2406-2415.
- Kim, B. (2017): Western Blot Techniques. Methods Mol. Biol., 1606, 133-139.
- Knierim, J.J. (2015): The hippocampus. Curr. Biol., 25, R1116-R1121.
- Li, H. (2017): TRP Channel Classification. Adv. Exp. Med. Biol., 976, 1-8.
- Liu, M., Liu, X., Wang, L., Wang, Y., Dong, F., Wu, J., Qu, X., Liu, Y., Liu, Z., Fan, H. and Yao, R. (2018): TRPV4 Inhibition Improved Myelination and Reduced Glia Reactivity and Inflammation in a Cuprizone-Induced Mouse Model of Demyelination. Front. Cell. Neurosci., 12, 392.
- Mion, G. and Villevieille, T. (2013): Ketamine pharmacology: an update (pharmacodynamics and molecular aspects, recent findings). CNS Neurosci. Ther., 19, 370-380.
- Mitchell, S., Vargas, J. and Hoffmann, A. (2016): Signaling via the NFκB system. Wiley Interdiscip. Rev. Syst. Biol. Med., 8, 227-241.
- Mizrak, A., Erbagci, I., Arici, T., Ozcan, I., Ganidagli, S., Tatar, G. and Oner, U. (2010): Ketamine versus propofol for strabismus surgery in children. Clin. Ophthalmol., 4, 673-679.
- Morgan, C.J. and Curran, H.V.; Independent Scientific Committee on Drugs. (2012): Ketamine use: a review. Addiction, 107, 27-38.
- Morty, R.E. and Kuebler, W.M. (2014): TRPV4: an exciting new target to promote alveolocapillary barrier function. Am. J. Physiol. Lung Cell. Mol. Physiol., 307, L817-L821.
- Neuhäuser, C., Wagner, B., Heckmann, M., Weigand, M.A. and Zimmer, K.P. (2010): Analgesia and sedation for painful interventions in children and adolescents. Dtsch. Arztebl. Int., 107, 241-247. I-II, I.
- Ohsaki, A., Tanuma, S.I. and Tsukimoto, M. (2018): TRPV4 Channel-Regulated ATP Release Contributes to γ-Irradiation-Induced Production of IL-6 and IL-8 in Epidermal Keratinocytes. Biol. Pharm. Bull., 41, 1620-1626.
- Owen, J.B. and Butterfield, D.A. (2010): Measurement of oxidized/reduced glutathione ratio. Methods Mol. Biol., 648, 269-277.
- Rame, M., Caudal, D., Schenker, E., Svenningsson, P., Spedding, M., Jay, T.M. and Godsil, B.P. (2017): Clozapine counteracts a ketamine-induced depression of hippocampal-prefrontal neuroplasticity and alters signaling pathway phosphorylation. PLoS One, 12, e0177036.
- Sies, H. (2015): Oxidative stress: a concept in redox biology and medicine. Redox Biol., 4, 180-183.
- Sinha, N. and Dabla, P.K. (2015): Oxidative stress and antioxidants in hypertension-a current review. Curr. Hypertens. Rev., 11, 132-142.
- Venkatachalam, K. and Montell, C. (2007): TRP channels. Annu. Rev. Biochem., 76, 387-417.
- Wei, C., Li, L., Kim, I.K., Sun, P. and Gupta, S. (2014): NF-κB mediated miR-21 regulation in cardiomyocytes apoptosis under oxidative stress. Free Radic. Res., 48, 282-291.
- Yassa, M.A. and Stark, C.E. (2011): Pattern separation in the hippocampus. Trends Neurosci., 34, 515-525.
- Ye, Z., Li, Q., Guo, Q., Xiong, Y., Guo, D., Yang, H. and Shu, Y. (2018): Ketamine induces hippocampal apoptosis through a mechanism associated with the caspase-1 dependent pyroptosis. Neuropharmacology, 128, 63-75.
- Zhang, L., Papadopoulos, P. and Hamel, E. (2013): Endothelial TRPV4 channels mediate dilation of cerebral arteries: impairment and recovery in cerebrovascular pathologies related to Alzheimer’s disease. Br. J. Pharmacol., 170, 661-670.
- Zhang, Q., Lenardo, M.J. and Baltimore, D. (2017): 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell, 168, 37-57.
- Zuo, T., Zhu, M., Xu, W., Wang, Z. and Song, H. (2017): Iridoids with Genipin Stem Nucleus Inhibit Lipopolysaccharide-Induced Inflammation and Oxidative Stress by Blocking the NF-κB Pathway in Polycystic Ovary Syndrome. Cell. Physiol. Biochem., 43, 1855-1865.