INTRODUCTION
Lysosomes are essential organelles responsible for the calcium signaling, nutrient responses, and cholesterol homeostasis in eukaryotic cells. These organelles are central for the degradation and recycling of intracellular and extracellular substances derived from autophagic and endocytic pathways, respectively. Lysosomes are single-membrane-bound organelles with acidic environments maintained by the V-ATPase proton pumps, chloride channels, and ion transporters in the lysosomal membrane and contain > 60 different acid hydrolases, including cathepsins and glucocerebrosidase (GCase). Maintaining the lysosomal integrity and function is important for cellular homeostasis (Appelqvist et al., 2013; Xu and Ren, 2015).
Lysosomes also play key roles in cell death. Low lysosomal pH and high lysosomal content of calcium and acid hydrolases can trigger lysosome-dependent cell death (LDCD). Lysosomal membrane permeabilization (LMP) leads to LDCD because of lysosomal dysfunction and leakage of lysosomal contents (Kroemer and Jäättelä, 2005; Boya and Kroemer, 2008). Recent studies have suggested that LMP is initiated by external stimuli, such as: lysosomotropic agents, including the leucine dipeptide L-leucyl–L-leucine methyl ester (LLOMe) (Thiele and Lipsky, 1990); particles, including silica; or pathogens, including bacteria (Thibodeau et al., 2004; Aits and Jäättelä, 2013). In addition, LMP is also induced by internal stimuli, such as: reactive oxygen species (Dehay et al., 2010; Denamur et al., 2011; Wu et al., 2015); neurotoxic aggregates, including α-synuclein, amyloid-β, mutant huntingtin, and tau fibrils (Flavin et al., 2017); or uric acid crystals: thereby exacerbating neurodegenerative diseases, such as Parkinson’s, Alzheimer’s, and Huntington’s disease, as well as kidney injury (Maejima et al., 2013).
Several studies have reported that lysosome repair and clearance pathways are activated in response to lysosomal damage. Minor damage to the lysosomal membrane is repaired by the endosomal sorting complexes required for transport (ESCRT) machinery upon the release of the lysosomal calcium to the cytosol (Radulovic et al., 2018; Skowyra et al., 2018). Severely damaged lysosomes are ubiquitinated to be cleared through macroautophagy, termed lysophagy (Papadopoulos and Meyer, 2017; Yoshida et al., 2017). These lysosomal repair and removal pathways that are activated after LMP maintain the lysosomal homeostasis. The expression of most lysosomal proteins, including lysosomal hydrolases (e.g., cathepsins A, B, and D, and GCase), and lysosomal membrane proteins (e.g., lysosome-associated membrane protein [LAMP]1, LAMP2, and mucolipin 1), is determined by the CLEAR (coordinated lysosomal expression and regulation) gene network. This network is regulated by the microphthalmia/transcription factor E (MiT/TFE) family, which includes transcription factor EB (TFEB), transcription factor E3 (TFE3), and microphthalmia-associated transcription factor (MITF). Under normal conditions, mammalian target of rapamycin (mTOR) keeps TFEB at the phosphorylated, inactive state. Upon lysosomal damage, mTOR complex 1 dissociates from the damaged lysosomes. TFEB are subsequently dephosphorylated and then translocate from the cytosol to the nucleus to activate lysosomal gene expression (Sardiello et al., 2009; Settembre et al., 2011; Napolitano and Ballabio, 2016). The same mechanisms that regulate TFEB activity also appear to control the activity of other MiT/TFE members (Martina and Puertollano, 2013; Martina et al., 2014; Napolitano and Ballabio, 2016). Although the activation of TFEB after lysosomal-membrane damage has been reported (Chauhan et al., 2016; Jia et al., 2018; Arhzaouy et al., 2019; Rusmini et al., 2019; Nakamura et al., 2020), it is not clear whether this response contributes to suppressing the cell death associated with lysosomal-membrane damage. Therefore, we investigated in HeLa cells the contribution of MiT/TFE family members to reducing the cell death associated with lysosomal-membrane damage induced by LLOMe.
MATERIALS AND METHODS
Reagents
LLOMe monohydrochloride (16008) was purchased from Cayman Chemical (Ann Arbor, MI, USA). Ethanol (054-07225), MgCl2∙6H2O (135-00165), Na3VO4 (198-09752), digitonin (043-21376), mouse anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (016-25523), sodium acetate (192-01075), and NaCl (191-01665) were purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). Dulbecco’s Modified Eagle’s Medium (DMEM; 08456-36), Penicillin-Streptomycin Mixed Solution (09367-34), HEPES (17514-15), KCl (28538-75), EDTA (15105-35), dithiothreitol (14128-04), protease inhibitor cocktail (25955-11), Nonidet P-40 (23640-94), glycerol (17045-65), 4% paraformaldehyde phosphate-buffered solution (PBS) (09154-14), Blocking One Histo (06349-64), Bullet Blocking One (13779-01), LB Agar, Miller (20069-65), ampicillin sodium salt (02739-74), Tryptone (35640-95), and Yeast Extract Dried (15838-45) were purchased from Nacalai Tesque, Inc. (Kyoto, Japan). Fetal Bovine Serum (FB-1285/500) was purchased from Biosera (Nuaille, France). Culture dishes (100 mm; 130182, 60 mm; 150462, 35 mm; 150460), Alexa Fluor 488-conjugated goat anti-mouse IgG (A11001), Alexa Fluor 555-conjugated goat anti-rabbit IgG (A31629), 4’,6-diamidino-2-phenylindole dihydrochloride (DAPI; D1306), Prolong Diamond Antifade Mountant (P36961), small interfering RNAs (siRNAs) (TFEB [s15495], TFE3 [s14031], MITF [s8790], and Negative Control No. 1 [4390843]), Lipofectamine RNAi MAX transfection Reagent (13778150), Opti-MEM I Reduced Serum Medium (31985070), Lipofectamine LTX Reagent (15338100), and pcDNA3.1/Zeo(+) Mammalian Expression Vector (V86020) were purchased from Thermo Fisher Scientific, Inc. (Waltham, MA, USA). Bio-Rad Protein Assay Dye Reagent Concentrate (500-0006) and Tween 20 (161-0781) were purchased from Bio-Rad Laboratories, Inc. (Hercules, CA, USA). Skim milk was purchased from Meiji Co., Ltd. (Tokyo, Japan). Can Get Signal Solution 1 (NKB-201), Solution 2 (NKB-301), Solution A (NKB-501), SuperPrep II/KOD SYBR qPCR Set (SCQ401/QKD201), KOD One PCR Master Mix -Blue- (KMM-201), Hind III (HND-311), and Ligation High Ver.2 (LGK-201) were purchased from TOYOBO CO., LTD. (Osaka, Japan). Rabbit anti-TFEB antibody (4240), rabbit anti-TFE3 antibody (14779), and rabbit anti-MITF antibody (12590) were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). Mouse anti-β-actin (M177-3) was purchased from MEDICAL & BIOLOGICAL LABORATORIES CO., LTD. (Aichi, Japan). Tris buffered saline (sc-362308) and mouse anti-Lamin B1 (sc-377000) were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). NaF (450022), horse radish peroxidase (HRP)-conjugated anti-rabbit IgG (A9169), and HRP-conjugated anti-mouse IgG (A9044) were purchased from Sigma-Aldrich Co LLC. (St Louis, MO, USA). Poly-D-lysine-coated 4-well chamber slides (354577) and 96-well microplates (353072) were purchased from Corning Incorporated (Corning, NY, USA). Mouse anti-Galectin-3 antibody (126701) was purchased from BioLegend, Inc. (San Diego, CA, USA). Cell Counting Kit-8 (CK04) and Cytotoxicity LDH Assay Kit-WST (CK12) were purchased from Dojindo Laboratories (Kumamoto, Japan). M-MLV Reverse Transcriptase (M170A) was purchased from Promega (Madison, WI, USA). FavorPrep GEL/PCR Purification Mini Kit (FAGCK001-1) and Plasmid DNA Extraction Mini kit (FAPDE 001-1) were purchased from Favorgen Biotech Corporation (Ping-Tung, Taiwan). Nuclease-free water (312-90103) and BamH I (315-00061) were purchased from NIPPON GENE CO., LTD. (Tokyo, Japan). SoloPack Gold Supercompetent Cells (230350) was purchased from Agilent Technologies, Inc. (Santa Clara, CA, USA). EmeraldAmp MAX PCR Master Mix (RR320A) was purchased from Takara Bio Inc. (Shiga, Japan).
Cell culture
The human cervical cancer cell line HeLa cells were cultured in DMEM with 10% Fetal Bovine Serum and 1% Penicillin-Streptomycin in a humidified 5%-CO2 incubator at 37°C. The medium was changed every 2 days, and the cells were passaged when they reached approximately 80% confluence. In all experiments, cells were seeded at a density of 4,651 cells/197 μL/cm2 and incubated overnight in the incubator before treated with vehicle control (0.3% ethanol) or LLOMe.
Immunocytochemical analysis
HeLa cells were seeded in poly-D-lysine-coated 4-well chamber slides and incubated overnight at 37°C in a humidified 5%-CO2 incubator. The cells were exposed to LLOMe as indicated, washed twice with PBS(−), and fixed with 4% paraformaldehyde PBS for 15 min at room temperature. The fixed cells were washed three times with PBS(−) and permeabilized with 100 μg/mL digitonin in PBS(−) for 10 min at room temperature. The cells were then washed three times with PBS(−) and blocked with Blocking One Histo for 15 min at room temperature. The cells were washed with PBS(−) and incubated overnight at 4°C with mouse anti-Galectin-3 (1:200) and anti-TFEB (1:400) antibodies diluted in Can Get Signal Solution A. The cells were then washed three times with PBS(−), incubated in the dark for 1 hr at room temperature with Alexa Fluor 488-conjugated goat anti-mouse IgG (1:800) and Alexa Fluor 555-conjugated goat anti-rabbit IgG (1:800) diluted in Can Get Signal Solution A, then washed three times with PBS(−), and incubated with 600 nM DAPI in PBS(−) for 5 min at room temperature in the dark. After a final wash with PBS(−), the cells were mounted using Prolong Diamond Antifade Mountant. After overnight incubation, the slides were observed under a confocal laser scanning microscope (FV1000-D, Olympus Corporation, Tokyo, Japan). Images were processed using the FV10-ASW 4.2 Viewer (Olympus Corporation), and whole-image contrast was adjusted using ImageJ software (http://imagej.nih.gov/ij/).
Nucleocytoplasmic fractionation
Nucleocytoplasmic fractionation was performed as described by Ishida et al. (2017), with minor modifications. HeLa cells were seeded in 100-mm dishes and incubated overnight at 37°C in a humidified 5%-CO2 incubator. After LLOMe exposure, the cells were harvested by trypsinization and washed with PBS(−). The cells were then centrifuged at 1,100 rpm for 3 min at 4°C, resuspended in 100 μL of hypotonic buffer [10 mM HEPES, pH 7.9, 1.5 mM MgCl2∙6H2O, 10 mM KCl, 0.1 mM EDTA, 1 mM NaF, 1 mM Na3VO4, 1 mM dithiothreitol, and 1% protease inhibitor cocktail], and incubated for 15 min on ice. Cell membranes were broken by the addition of 5 μL of 10% Nonidet P-40, followed by vortexing. The cells were centrifuged at 10,000 × g for 5 min at 4°C, and the supernatants were collected as the cytoplasmic fractions. The pellets were washed twice by resuspending in the hypotonic buffer, vortexing, and then centrifugating at 10,000 × g for 5 min at 4°C. The pellets were finally resuspended in 50 μL of high-salt extraction buffer [20 mM HEPES, pH 7.9, 1.5 mM MgCl2∙6H2O, 400 mM KCl, 0.1 mM EDTA, 10% glycerol, 1 mM NaF, 1 mM Na3VO4, 1 mM dithiothreitol, and 1% protease inhibitor cocktail], sonicated on ice, and rotated for 30 min at 4°C. The supernatants were collected as the nuclear fractions. Total protein content in the cytoplasmic and nuclear fractions were quantified using the Bio-Rad Protein Assay Dye Reagent Concentrate.
Western blotting
Western blotting was performed as described by Miyara et al. (2016b) and Umeda et al. (2016) with minor modifications. In this study, an equivalent amount of total protein (5 μg) from each sample was loaded onto the gels. Membranes were blocked at room temperature for 1 hr with 5% skim milk in tris-buffered saline containing 0.1% tween 20 (TBST) or for 5 min with Bullet Blocking One. They were then washed three times with TBST and probed overnight at 4°C with the following antibodies diluted in Can Get Signal Solution 1: rabbit anti-TFEB (1:2,000), rabbit anti-TFE3 (1:2,000), rabbit anti-MITF (1:2,000), mouse anti-GAPDH (1:8,000), mouse anti-Lamin B1 (1:2,000) and mouse anti-β-actin (1:10,000). Membrane-bound primary antibodies were detected by incubating the membranes for 1 hr at room temperature with the following HRP-labeled secondary antibodies diluted in Can Get Signal Solution 2: HRP-conjugated anti-rabbit IgG (1:8,000), and HRP-conjugated anti-mouse IgG (1: 8,000 or 1:20,000 or 1:100,000). Chemiluminescent signals were detected using a luminescent image analyzer (FUSION SOLO.7S.EDGE, Vilber Lourmat, Marne-la-Vallée, France), and band intensities were quantified via densitometric analysis using Evolution-Capt software (Vilber Lourmat).
Whole-cell protein extraction
HeLa cells were seeded in 60-mm dishes. Whole-cell protein extraction was performed using method 1 described by Miyara et al. (2016a).
Cell lysis, reverse transcription, and real-time PCR
HeLa cells were seeded in 96-well microplates. Cell lysis, reverse transcription, and real-time PCR were performed using the SuperPrep II/KOD SYBR qPCR Set and PikoReal 96 Real-Time PCR System (TCR0096, Thermo Fisher Scientific, Inc., Waltham, MA, USA) according to the manufacturer’s instructions. The sequences of the primers used are shown in Table 1. The quantification cycle values were determined using PikoReal Software 2.1 (Thermo Fisher Scientific, Inc.), and the relative changes in gene expression were calculated by using the ΔΔCt method with RPL13A used as the reference gene.
Table 1. The sequences of the primers used in real-time RT-PCR analysis.
| Gene |
Primer |
Sequence (5'-3') |
| CTSA |
Forward |
5'-CAGGCTTTGGTCTTCTCTCCA-3' |
| Reverse |
5'-TCACGCATTCCAGGTCTTTG-3' |
| CTSB |
Forward |
5'-AGTGGAGAATGGCACACCCTA-3' |
| Reverse |
5'-AAGAAGCCATTGTCACCCCA-3' |
| CTSD |
Forward |
5'-CTGATTCAGGGCGAGTACATGA-3' |
| Reverse |
5'-CGACACCTTGAGCGTGTAGT-3' |
| GBA |
Forward |
5'-TGGGTACCCGGATGATGTTA-3' |
| Reverse |
5'-AGATGCTGCTGCTCTCAACA-3' |
| LAMP1 |
Forward |
5'-ACGTTACAGCGTCCAGCTCAT-3' |
| Reverse |
5'-TCTTTGGAGCTCGCATTGG-3' |
| LAMP2 |
Forward |
5'-ACACAACATTTCCTGATGCTGA-3' |
| Reverse |
5'-TTTGTGCTCACTGTGCCATTT-3' |
| MCOLN1 |
Forward |
5'-TTGCTCTCTGCCAGCGGTACTA-3' |
| Reverse |
5'-GCAGTCAGTAACCACCATCGGA-3' |
| RPL13A |
Forward |
5'-GGAGGAGGAGAGGAAAGAGA-3' |
| Reverse |
5'-TTGAGGACCTCTGTGTATTTGTCAA-3' |
RNA interference
HeLa cells were seeded at a density of 1 × 105 cells in 35-mm dishes and incubated overnight at 37°C in a humidified 5%-CO2 incubator. The cells were then transfected with 15 nM (5 nM each) of the Silencer Select Pre-designed siRNAs by using Lipofectamine RNAi MAX transfection Reagent according to the manufacturer’s instructions. Afterward, the cells were incubated for 24 hr in the incubator and then reseeded in 60-mm dishes or 96-well microplates for subsequent experiments.
Viability/Cytotoxicity assay
HeLa cells were seeded in 96-well microplates. Cell viability was measured using Cell Counting Kit-8. After LLOMe exposure, the medium of the culture was collected for cytotoxicity assay and replaced with 50 μL of Cell Counting Kit-8 diluted 1: 4 in fresh culture medium. After incubation at 37°C in a humidified 5%-CO2 incubator for 30 min, the absorbance of the converted dye was measured at 450 nm by using a MultiSkan Go microplate spectrophotometer (Thermo Fisher Scientific, Inc.). Cytotoxicity was measured using Cytotoxicity LDH Assay Kit-WST. After LLOMe exposure, 50 μL of the medium of the culture diluted 1: 1 in fresh culture medium was mixed with 50 μL of Working Solution. After incubation at 37°C for 30 min in the dark, 25 μL of Stop Solution was added, and the absorbance of the converted dye was measured at 490 nm by using the microplate spectrophotometer. Maximal LDH was released from the cells into the medium by adding 10 μL of Lysis Solution and then incubating the cells at 37°C in a humidified 5%-CO2 incubator for 30 min.
cDNA synthesis and plasmid construction
First-stand cDNA synthesis from HeLa-cell total RNA was performed using M-MLV Reverse Transcriptase according to the manufacturer’s instructions. The cDNA fragment encoding human influenza hemagglutinin (HA)-tagged human TFEB was amplified via PCR using KOD One PCR Master Mix -Blue- according to the manufacturer’s instructions. The PCR amplicons were resolved via electrophoresis and purified using the FavorPrep GEL/PCR Purification Mini Kit according to the manufacturer’s instructions. Purified cDNA was further amplified via a second round PCR. The PCR amplicon was precipitated using 99.5% ethanol and 3 M sodium acetate (pH 5.2) and resuspended in nuclease-free water. The cDNA fragment (2 μg) and pcDNA3.1/Zeo(+) Mammalian Expression Vector (2 μg) were cleaved with Hind III and BamH I according to the manufacturer’s instructions. The digestion products were resolved and purified as described above. The cDNA fragment was ligated to pcDNA3.1/Zeo(+) Mammalian Expression Vector by using Ligation High Ver.2 according to the manufacturer’s instructions. The plasmid construct was mixed with SoloPack Gold Supercompetent Cells, which were then incubated on ice for 15 min and induced to acquire the plasmid by incubating them at 42°C for 40 sec, and then on ice for 3 min. The cells were then plated on Luria Broth agar plates (4% LB Agar, Miller) containing 50 μg/mL ampicillin sodium salt and incubated overnight at 37°C. Colonies were individually picked and the insert cDNA fragment was amplified via direct colony PCR using EmeraldAmp MAX PCR Master Mix according to the manufacturer’s instructions. A PCR-positive colony was suspended in 2 × Yeast Extract Tryptone medium (1.6% Tryptone, 1% Yeast Extract Dried, and 0.5% NaCl; pH 7) containing 50 μg/mL ampicillin sodium salt and incubated overnight at 37°C while shaking at 250 rpm. The plasmid construct was purified using the Plasmid DNA Extraction Mini kit according to the manufacturer’s instructions. The insert cDNA sequence was determined via Sanger DNA sequencing (performed by GENEWIZ Japan Corp., Saitama, Japan). The sequences of the primers used are shown in Table 2.
Table 2. The sequences of primers used in HA-TFEB expression plasmid construction.
| Method |
Primer |
Sequence (5'-3') |
| 1st PCR |
Forward1 |
5'-TACCCATACGATGTTCCAGATTACGCTATGGCGTCACGCATAGGG-3' |
| Reverse2 |
5'-TGATATCATAGGATCCTCACAGCACATCGCCCTCCTC-3' |
| 2nd PCR |
Forward3 |
5'-GTACGAGACGAAGCTTGCACCATGTACCCATACGATGTTCCAGA-3' |
| Reverse2 |
5'-TGATATCATAGGATCCTCACAGCACATCGCCCTCCTC-3' |
| Colony-direct PCR |
Forward |
5'-CGCAAATGGGCGGTAGGCGTG-3' |
| Reverse |
5'-TGATATCATAGGATCCTCACAGCACATCGCCCTCCTC-3' |
| Sanger DNA sequencing |
Forward |
5'-CGCAAATGGGCGGTAGGCGTG-3' |
| Forward |
5'-GGAACAAGTTTGCTGCCCAC-3' |
| Forward |
5'-TCTGTGGATT ACATCCGGAG-3' |
containing: 1HA tag coding sequence, 2BamH I restriction site, 3Hind III restriction site and the ATG start codon.
Plasmid transfection
HeLa cells were seeded at a density of 1 × 105 cells in 35-mm dishes and incubated overnight at 37°C in a humidified 5%-CO2 incubator. The cells were then transfected with plasmid constructs by using Lipofectamine LTX Reagent. Briefly, each plasmid construct (1.25 μg) was mixed with 500 μL of Opti-MEM I Reduced Serum Medium and 3.75 μL of Lipofectamine LTX Reagent, vortexed, and incubated for 25 min at room temperature. Afterward, the medium of the culture was changed, and the plasmid construct/Lipofectamine LTX Reagent mixture was added to the culture. The cells were incubated for 24 hr and then re-seeded in 60-mm dishes or 96-well microplates for subsequent experiments.
Statistics
Data are expressed as the mean ± SD from at least three independent experiments. The significance of the differences between two independent groups was determined according to Student’s t-test by using Mini-StatMate software (ATMS Co. Ltd., Tokyo, Japan). The significance of the differences among multiple independent groups was determined according to one-way ANOVA followed by Tukey’s post-hoc test by using Mini-StatMate software (ATMS Co. Ltd.). A P-value of < 0.05 was considered statistically significant.
RESULTS
LLOMe activates MiT/TFE family members and increases the mRNA levels of their target genes
Galectin-3 is a β-galactoside-binding lectin normally found in both the cytoplasm and nucleus and recognizes the exposed glycoproteins on the luminal side of a damaged lysosomal membrane, thereby acting as a sensor of lysosomal damage (Aits et al., 2015). To temporally investigate the lysosomal damage and TFEB nuclear translocation induced by LLOMe, we treated HeLa cells with ethanol or 1 mM LLOMe for the indicated time and evaluated the subcellular localization of Galectin-3 and TFEB via immunocytochemical analysis. We observed that 1 mM LLOMe markedly induced the formation of Galectin-3 puncta and TFEB nuclear translocation after 6 and 24 hr (Fig. 1). Additionally, TFEB colocalized with the Galectin-3 puncta (Fig. 1). The number of cells with TFEB nuclear localization was decreased 48 hr after exposure to 1 mM LLOMe, coincident with a decrease in the number of intracellular Galectin-3 puncta (Fig. 1). We also examined the temporal changes in the nuclear translocation of TFEB and two other MiT/TFE family members, TFE3 and MITF, via Western blotting analysis and observed that the nuclear translocation of all the three members increased after 1 hr of 1 mM LLOMe treatment and then gradually returned to the control levels over 48 hr (Fig. 2). To confirm whether LLOMe-induced nuclear translocation of MiT/TFE family members can indeed upregulate their target genes, we exposed HeLa cells to ethanol or 1 mM LLOMe for 6, 24, or 48 hr, before measuring the mRNA expression levels of TFEB/TFE3/MITF target genes via real-time reverse transcription (RT)-PCR. We observed that 1 mM LLOMe increased the mRNA levels of genes encoding lysosomal hydrolases CTSA, CTSB, CTSD, and GBA after 24 hr (Figs. 3a-d). Likewise, the mRNA levels of genes encoding lysosomal membrane proteins LAMP1, LAMP2, and MCOLN1 were also increased after 24 hr (Figs. 3e-g). These increases in the mRNA levels of TFEB/TFE3/MITF target genes were maintained or decreased after 48 hr (Fig. 3). These results suggest that lysosomal-membrane damage induced by LLOMe rapidly activates MiT/TFE family members, whereby their target genes are eventually upregulated.

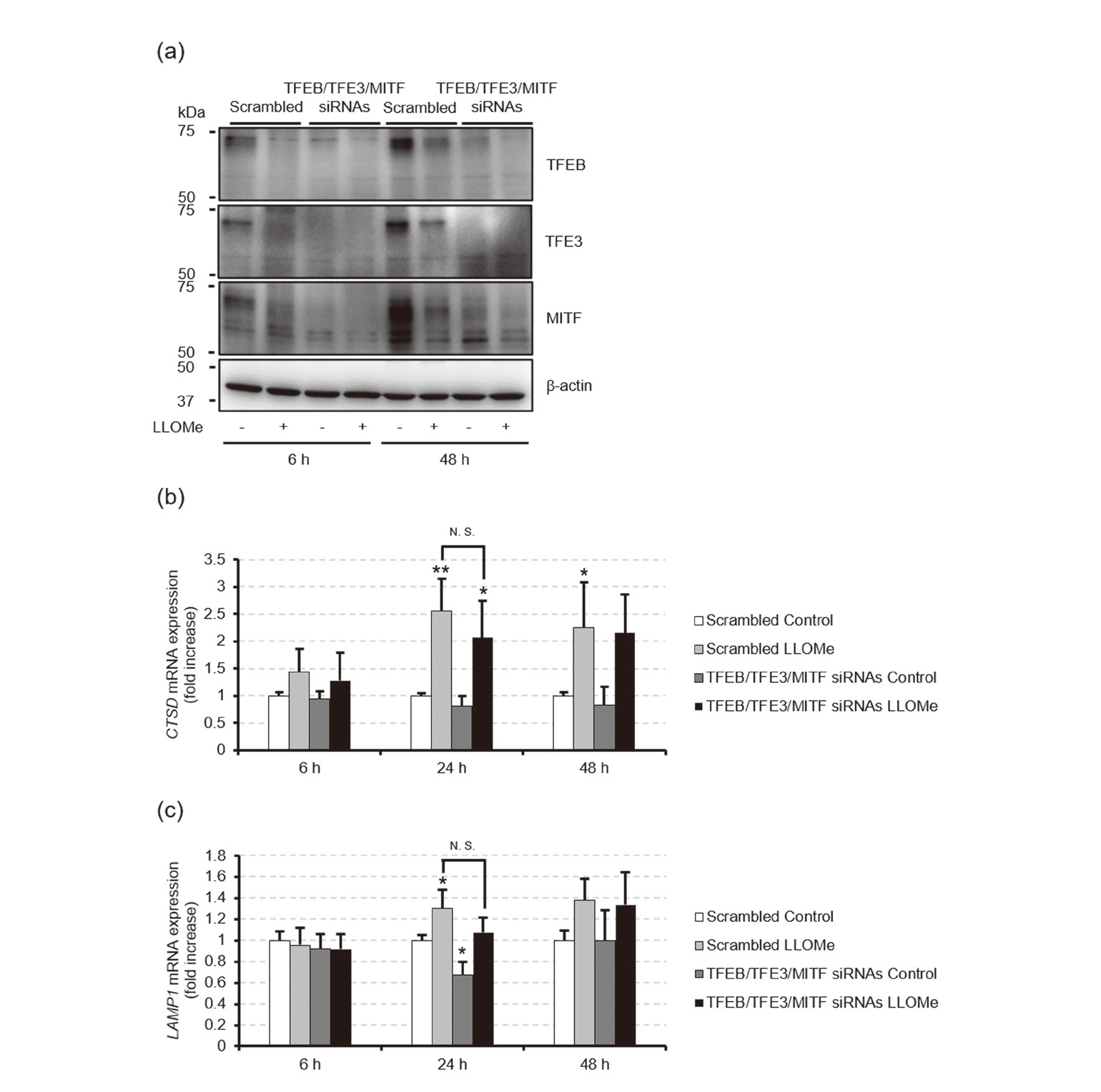
To assess whether the lysosomal biogenesis mediated by MiT/TFE family members contributes to suppressing LLOMe-induced cell death, we exposed HeLa cells transfected with a scrambled control siRNA or siRNAs specific for TFEB/TFE3/MITF to ethanol or 1 mM LLOMe for 6, 24, or 48 hr and measured cell viability and cytotoxicity by using commercial assay kits. Western blotting and real-time RT-PCR analyses revealed that the siRNAs decreased the TFEB/TFE3/MITF protein levels (Fig. 4a), whereby the increases in CTSD and LAMP1 mRNA levels induced by 24-hr exposure to 1 mM LLOMe were slightly inhibited, from 2.5 ± 0.59-fold to 2.1 ± 0.67-fold of scrambled siRNA-transfected control and from 1.3 ± 0.18-fold to 1.1 ± 0.14-fold of scrambled siRNA-transfected control, respectively, although they were not statistically significant (Figs. 4b and c). Additionally, the siRNA-mediated knockdown of TFEB/TFE3/MITF exacerbated cell death induced by 24 or 48 hr of 1 mM LLOMe exposure, as evidenced by the decrease in cell viability concomitant with the increase in the release of the cytoplasmic enzyme lactate dehydrogenase (LDH) into the medium from the damaged cells, but these effects were not observed upon 6 hr of LLOMe exposure (Fig. 5). These results suggest that MiT/TFE family members contribute to suppressing the LLOMe-induced cell death.

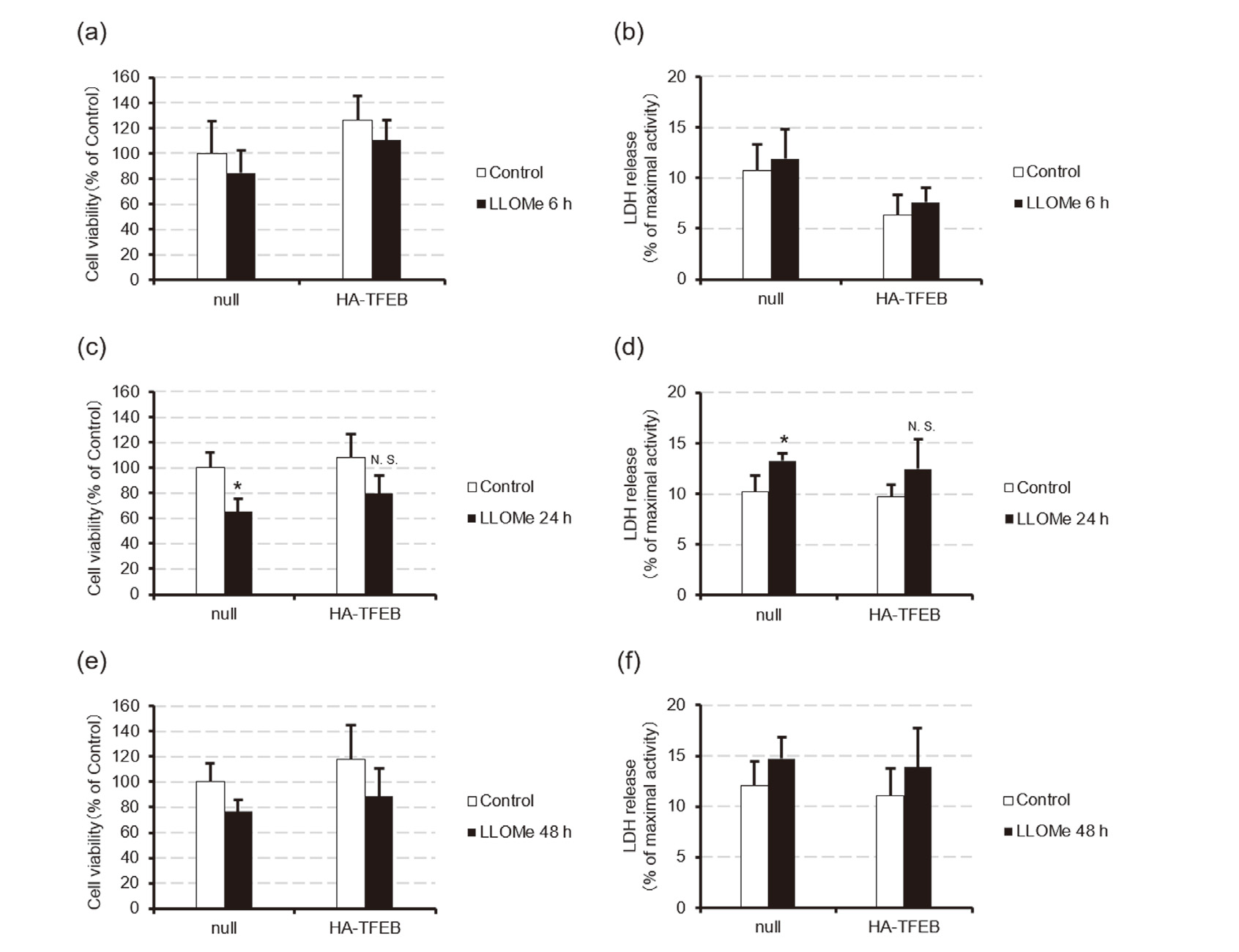
To examine whether enhancement of TFEB-mediated lysosomal biogenesis attenuates LLOMe-induced cell death, we exposed HeLa cells transfected with an HA-TFEB expression plasmid or the null-plasmid to ethanol or 1 mM LLOMe for 6, 24, or 48 hr and measured the cell viability and LDH release. Western blotting analysis revealed that HA-TFEB expression plasmid effectively increased the TFEB protein levels, which gradually decreased over 48 hr (Fig. 6a). Additionally, LLOMe decreased the levels of TFEB protein soluble in 1% Nonidet P-40, presumably due to the nuclear translocation of the protein (Fig. 6a). Consistent with these observations, TFEB overexpression significantly enhanced the increase in CTSD mRNA levels induced by 24-hr exposure to1 mM LLOMe, from 2.2 ± 0.64-fold to 3.4 ± 0.52-fold of null plasmid-transfected control, although this effect was not observed in LAMP1 mRNA levels (Figs. 6b and c). However, TFEB overexpression only slightly attenuated the LLOMe-induced decrease in cell viability and increase in LDH release (Fig. 7). These results suggest that genetic enhancement of the TFEB-mediated lysosomal biogenesis has little effect in preventing LLOMe-induced cell death.

DISCUSSION
A recent study suggests that lysosomal damage induced by LLOMe stimulates lysosomal biogenesis via activation of TFEB, contributing to lysophagic clearance of damaged lysosomes (Nakamura et al., 2020). However, whether the lysosomal biogenesis mediated by MiT/TFE family members exerts a protective role against LLOMe-induced toxicity is not well studied. In this study, we demonstrated that LLOMe rapidly activates TFEB/TFE3/MITF within 1 hr in HeLa cells, leading to upregulation of their target genes after 24 hr (Figs. 1-3). Furthermore, siRNA-mediated knockdown of TFEB/TFE3/MITF exacerbated cell death induced by 24-hr or 48-hr exposure to LLOMe (Figs. 4 and 5), suggesting that MiT/TFE family members contribute to suppressing the LLOMe-induced cell death. However, TFEB overexpression only slightly attenuated LLOMe-induced cell death, despite enhanced LLOMe-induced increase in CTSD mRNA levels (Figs. 6 and 7). Thus, the endogenous levels of MiT/TFE family members might be sufficient to promote lysosomal biogenesis in response to lysosomal damage, although future studies should address whether TFEB/TFE3/MITF overexpression is effective in preventing LLOMe-induced cell death.
Although LLOMe upregulated the mRNA levels of all the TFEB/TFE3/MITF target genes analyzed, the increase in LAMP1 and LAMP2 mRNA levels were relatively less than the increase in the mRNA levels of other target genes (Fig. 3). A recent study has shown that lysosomal damage induced by LLOMe generates small membrane vesicles containing lysosomal membrane proteins, such as LAMP1 and LAMP2, possibly for recycling such lysosomal proteins (Eriksson et al., 2020). The same study has also shown that protein synthesis of LAMP2 is not upregulated during lysosomal membrane repair (Eriksson et al., 2020). Thus, genes encoding lysosomal membrane proteins may exhibit lower sensitivity to lysosomal damage than those of lysosomal hydrolases.
siRNA-mediated knockdown of TFEB/TFE3/MITF only slightly inhibited the increases in CTSD and LAMP1 mRNA levels induced by 1 mM LLOMe (Figs. 4 and 5). Recent studies have found other transcriptional regulators of lysosomal biogenesis, such as bromodomain containing protein 4 (BRD4) and signal transducer and activator of transcription 3 (STAT3) (Sakamaki et al., 2017; Martínez-Fábregas et al., 2018; Ballabio and Bonifacino, 2020). Thus, these other transcriptional regulators might also be involved in the lysosomal biogenesis in response to lysosomal-membrane damage. TFEB/TFE3/MITF knockdown also slightly exacerbated cell death induced by 1 mM LLOMe (Figs. 4 and 5). A recent study has reported that 1 mM LLOMe stimulates ESCRT-mediated lysosomal membrane repair and lysophagy (Jia et al., 2020). Thus, under our experimental condition, most damaged lysosomes may be sufficiently repaired by the ESCRT machinery or delivered to remaining intact lysosomes via lysophagy for degradation. In this context, the lysosomal biogenesis mediated by MiT/TFE family members may play a major role in restoring cellular lysosomal content after lysosomal repair and removal, even though this biogenesis pathway partially contributes to suppressing the LLOMe-induced cell death.
Accumulating evidence suggests that enhancement of the lysosomal biogenesis mediated by MiT/TFE family members is an attractive therapeutic target for several human diseases associated with lysosomal dysfunction, such as lysosomal storage diseases and neurodegenerative diseases (Settembre et al., 2013; Moors et al., 2017). In accordance, the present study demonstrated that the lysosomal biogenesis mediated by MiT/TFE family members, at least partially, plays a protective role against cell death associated with lysosomal-membrane damage. However, the present study also implied that genetic enhancement of the lysosomal biogenesis mediated by MiT/TFE family members has little effect in preventing the cell death associated with lysosomal-membrane damage. Future studies should address whether enhancing the lysosomal biogenesis mediated by MiT/TFE family members can be effective in reducing cell death associated with other LMP-inducing stimuli.