Abstract
Liver malignant tumors (LMTs) have recently been reported as severe and life-threatening adverse drug events associated with drug-induced liver injury (DILI). DILIs are the most common adverse drug event and can cause the withdrawal of medicinal products or major regulatory action. To reduce the attrition rate and cost of drug discovery, various quantitative structure–toxicity relationship models have been proposed to predict the probability of a DILI based on the chemical structure of a drug. However, there are many unresolved issues regarding the predictors of LMT-inducing drugs, and biologically interpretable prediction models for LMT have not been developed. Here, we constructed prediction models for whether a drug is LMT-inducing based on the activity of molecular initiating events (MIEs), which are biologically interpretable features and are defined as the initial interaction between a molecule and biosystem. We then constructed five machine learning models (i.e., LightGBM, XGBoost, random forest, neural network, and support vector machine) and evaluated their predictive performances. LightGBM achieved the best performance among the tested models. The MIEs making the highest contribution to the model construction for drug-induced LMT were inducement of Enhanced Level of Genome Instability Gene 1 (human ATAD5), nuclear factor-κ B, and activation of thyrotropin-releasing hormone receptor. These results support the previous literature and can be related to the mechanism onset of drug-induced LMT. Our findings may provide useful knowledge for drug development, research, and regulatory decision-making and will contribute to building more accurate and meaningful DILI prediction models by increasing understanding of biological predictors.
INTRODUCTION
A drug-induced liver injury (DILI) is the most common adverse drug event (ADE) and can cause medicinal product withdrawal or major regulatory action (Onakpoya et al., 2016). Therefore, early DILI prediction may be an effective strategy for reducing the attrition rate and cost of drug discovery. However, such prediction is difficult because of the diverse clinical conditions and chemicals that can cause DILIs.
Recently, liver malignant tumors (LMTs), one of the most serious forms of DILI, have been reported in association with the use of steroids (e.g., androgenic anabolic steroids and oral contraceptives) (Woodward et al., 2019; Hardt et al., 2012; Solbach et al., 2015; Stoot et al., 2010), despite having passed rigorous tests for genotoxic carcinogens such as genotoxicity and mutagenicity tests in non-clinical studies. LMT-inducing drugs must be detected inherently in non-clinical studies, because they lead to significant decrease in patient’s quality of life by destroying their liver reserves. Therefore, drug-induced LMT is among the most important endpoints of ADE prediction.
Steroids represented in reports of LMTs are typical endocrine-disrupting chemicals (EDCs). EDCs interfere with the endocrine system and stress response by interacting with nuclear receptors (NRs) and stress response pathways (SRs) to engender myriad adverse developmental, reproductive, and immunological effects in organism (Diamanti-Kandarakis et al., 2009). Recently, several studies have shown an association between EDC-induced modulation of NRs/SRs and carcinogenesis (Safe et al., 2013; Namani et al., 2014; Mizushima and Miyamoto, 2019). Therefore, LMT-inducing drugs might share the carcinogenic mode of action of EDCs.
However, prediction models that can predict LMT-inducing drugs for humans and provide their upstream chemical–biological interactions remain largely undeveloped, for the following reasons: 1) Benchmark datasets for DILI based on clinical data, such as US-FDA’s Liver Toxicity Knowledge Base or DILIrank (Chen et al., 2016), are not annotated with the phenotypes of DILI, so a dataset of drug-induced LMTs in humans based on clinical data must be acquired using an alternative approach; 2) the omics data of the information related to chemical–biological interactions upstream of drug-induced LMT are missing. To construct a prediction model that can interpret biological aspects, we must overcome the missing data problem.
In a quantitative structure–toxicity relationship (QSTR) prediction for DILIs, Zhu and Kruhlak (2014) showed the validity of drug annotation by signal detection using the FDA Adverse Events Reporting System (FAERS) database. The drug annotation method using FAERS is a scalable approach that is applicable to other ADE annotations, and can therefore be utilized for annotating LMT-inducing drugs.
Many DILI prediction models rely on the QSTR methodology (Wang et al., 2019; He et al., 2019; Hong et al., 2017), but QSTR cannot clarify the biological aspects contributing to the prediction, even though multiple biological events occur between chemical exposure and DILI expression. To fill the gaps in the omics data, such as the chemical–biological interactions from chemical exposures to adverse outcomes, researchers have proposed the use of in silico tools. Gadaleta et al. (2018) showed that the molecular initiating event (MIE) activities predicted by QSTR models could be input as variables in a subsequent DILI prediction, thus achieving multi-step prediction of chemical-facilitated steatosis. The chemical–protein interactions approach (Amano et al., 2020) and toxicogenomic approach (Liu et al., 2020; Aguirre-Plans et al., 2021) are also valid for multi-step ADE prediction, which have high similarly to the attempt by Gadaleta et al. (2018) in mediating biological intermediate variables between exposure of chemical structure and adverse outcome. Therefore, the MIE activities predicted from chemical structures and their experimental values could be applied to the prediction of drug-induced LMT. In addition, using the compound-MIE interactions as input variables, which are applied QSTR models as complementary tools, would provide an exhaustive interpretation of the biological factors contributing to the prediction.
Previously, we examined LMT-inducing drugs in the FAERS database and detected that NRs/SRs modulation could be potentially MIE-specific to LMT-inducing drugs (Kurosaki and Uesawa, 2021). Hence, NRs/SRs activities as MIEs upstream of an adverse outcome can characterize the chemical–biological interactions in drug-induced LMT predictions.
In this study, we hypothesized that whether a drug may induce LMTs in humans can be predicted from the features related to MIE activities, which can be calculated from chemical structures. Based on this hypothesis and using the FAERS database, we attempted to develop MIE-based prediction models and evaluate factors as predictors of LMT-inducing drugs. To our knowledge, we show the first successful machine learning application of post-marketing data and MIE activities to the prediction of drug-induced LMTs in humans.
MATERIALS AND METHODS
Dataset
We used a dataset constructed in our previous study (Kurosaki and Uesawa, 2021) consisting of 111 drugs from the FAERS database that were classified as LMT-positive or -negative. As the predictions of platinum preparation and polymer compounds were not supported by QSTR models implemented on Toxicity Predictor (detailed in the section “Curation and prediction of MIE activities”), they were excluded from the dataset used in this study (n = 107; see Supplemental Information: SI. 1) This dataset contains 44 (41%) LMT-positive drugs and 63 (59%) LMT-negative drugs.
Outline of MIEs
The MIEs used in this study exert agonist and antagonist effects not only on the NR superfamily (including NR subfamily 1, e.g., thyroid hormone receptors, retinoic acid receptors, peroxisome proliferator-activated receptors, and vitamin D receptors), but also on NRs associated with xenobiotic metabolism such as aryl hydrocarbon receptor and the constitutive androstane receptor. We also adopted up to 59 MIEs, adding agonist and antagonist effects on SRs. As examples, we included an antioxidant response element involved in the intrinsic mechanism of defense against oxidative stress, the mitochondrial membrane potential as a parameter of mitochondrial function, and p53, which is a biomarker for DNA damage and other cellular stresses (see Supplemental Information: Table S1).
Curation and prediction of MIE activities
Toxicity Predictor was used to collect experimental results on the MIE activities of the drugs (n = 107) (Kurosaki et al., 2020). This platform provides the results of toxicity assays conducted during the Toxicology in the 21st Century (Tox21) Project and predicts toxicity according to QSTR models developed with the Tox21 database. For drugs without experimental results on their MIE activities, the probability of MIE activities was predicted with QSTR models in Toxicity Predictor (Fig. 1). Thereby, we could obtain the complemented MIE activities matrix of any group of compounds based on their chemical structure alone. The predicted probabilities were normalized such that the cutoff value obtained using the Youden index (Fawcett, 2006) was 0.5. Toxicity Predictor has two QSTR models with two different PubChem activity criteria (≥ 1 active or ≥ 40 active) for 59 endpoints (Kurosaki et al., 2020). Therefore, we predicted the probabilities of 118 MIEs for each drug and adopted these MIEs as the LMT prediction features. Among 107 drugs, the QSTR models predicted MIE activities for a maximum of 72 (67%) and a minimum of 14 (13%) drugs per column, and 66% of the data matrix included the predicted MIE features (see Supplemental Information: SI. 2).

We formulated the LMT prediction as a binary classification task and considered five machine learning (ML) algorithms: LightGBM (LGB: by LightGBM library), XGBoost (XGB: by XGBoost library), random forest (RF: by scikit-learn library), neural networks (NNs: implemented by Keras library), and support vector machine classifier (SVC) using the scikit-learn library. The modeling scheme was designed to integrate nested cross-validation (CV) with outer fivefold and inner fivefold, a Bayesian optimization (implemented by Optuna library) search for hyperparameters in inner loop of nested CV, and a multiple model ensemble averaging strategy. Nested CV was used to reduce the evaluation bias of the model performance and provide a more robust means than single CV for assessing the model’s transferability to unseen data (Cawley and Talbot, 2010). Bayesian optimization is recommended for hyperparameter tuning because it achieves better results for a test set while requiring fewer iterations than grid search and random search (Bergstra et al., 2013). Finally, we grouped different models together to maximize the prediction performance and robustness using the multiple model ensemble averaging strategy. All ML modeling processes were implemented in Python version 3.8.5.
Hyperparameter optimization
We conducted a fivefold CV for hyperparameter optimization in the inner loop by iterating the Bayesian optimization 60 times. The search space for the hyperparameters was predefined (see Supplemental Information: Table S2), and optimal hyperparameters that minimize the average log loss were detected.
Multiple model ensemble averaging
To predict the test sets of outer loops, the models constructed in the inner CV were integrated by the ensemble averaging method. Five different models were constructed based on five different training sets generated in an inner CV, and the prediction probabilities of a test set were calculated by averaging the prediction probabilities by the five models constructed in the inner CV. Note that the learning rate for the training process was set to 10−4 for all models. Other fixed hyperparameters can be accessed in the Supplemental Information (Table S2).
Evaluation metrics
The predictive performances of test sets in the outer loops were evaluated from confusion matrices, which included the number of true positives (TPs) (i.e., drugs correctly identified as positive), true negatives (TNs) (i.e., drugs correctly identified as negative), false negatives (FNs) (i.e., positive drugs incorrectly classified as negative), and false positives (FPs) (i.e., negative drugs incorrectly classified as positive). Six evaluation indices were used to evaluate the classification models:
1. The receiver operating characteristic–area under the curve (ROC–AUC) graphs the performance of a classification model at all thresholds. This curve plots two parameters: the sensitivity (SE) and specificity (SP).
2. The accuracy (ACC) is defined as the number of correctly predicted samples divided by the total number of samples:
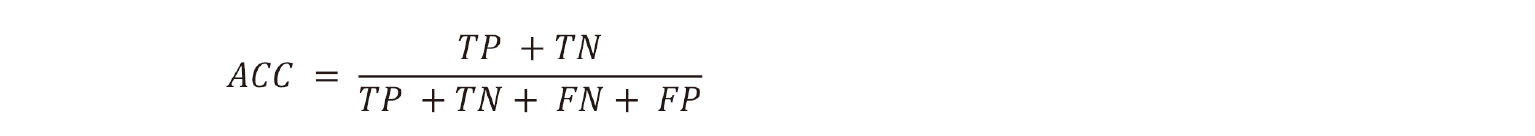
3. The SE is defined as the prediction accuracy when the true outcome is positive:

4. The SP is defined as the prediction accuracy when the true outcome is negative:

5. The balanced accuracy (BAC) is the average between the SE and SP:

6. The Matthews correlation coefficient (MCC) measures the classification accuracy of models for an unbalanced dataset (Matthews, 1975):
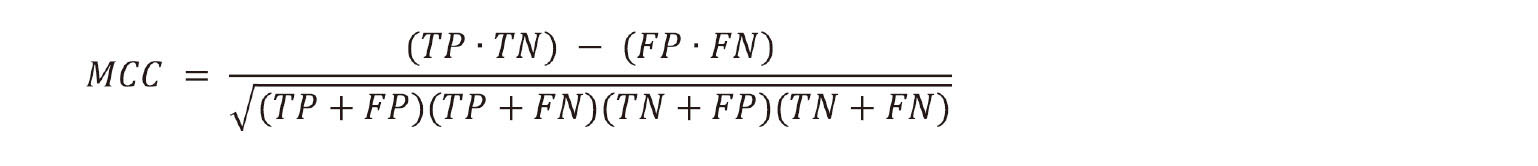
To determine the optimal cutoff point, (ratio of TP − ratio of FP) was maximized using the Youden index (Fawcett, 2006) for each test set prediction of the fivefold CV in the inner loop. By changing the value of the random number seed governing the fold division pattern, we generated 500 cutoff points and adopted their average value as the optimal cutoff point.
y-randomization
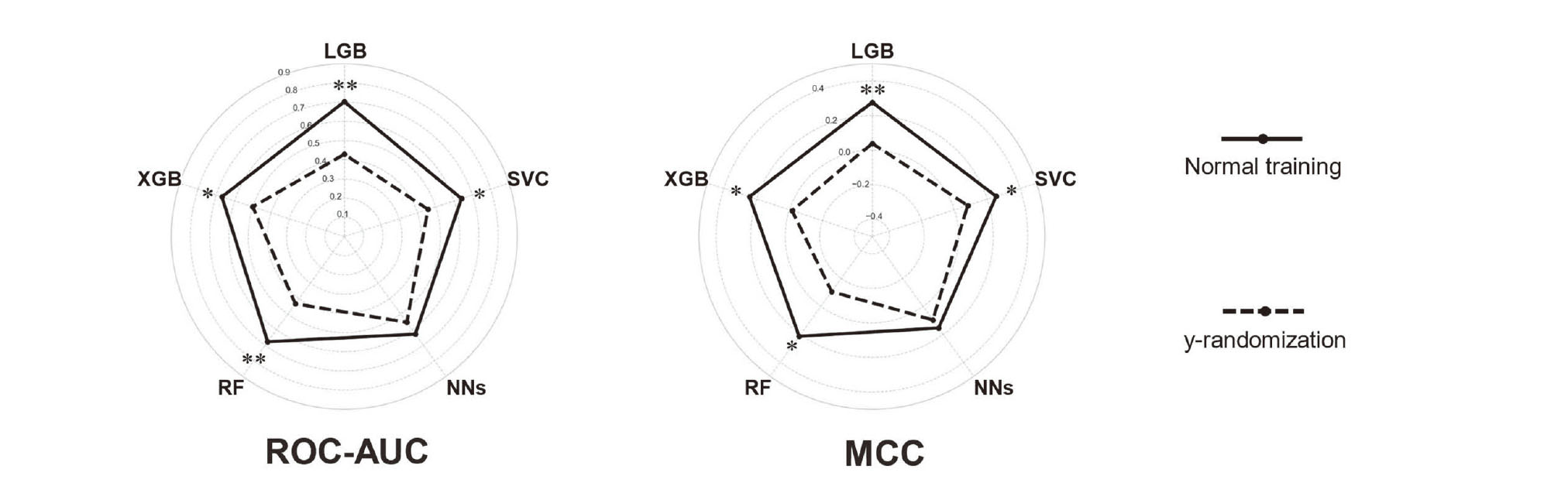
We performed y-randomization to evaluate the possibility of chance correlation in the constructed models. Objective variables were randomly assigned, and prediction models were trained, tested, and evaluated again. In the absence of chance correlation, the performances of the randomized models should decrease. We evaluated the possibility of chance correlation according to the ROC–AUC and MCC.
Evaluation of contributing MIEs
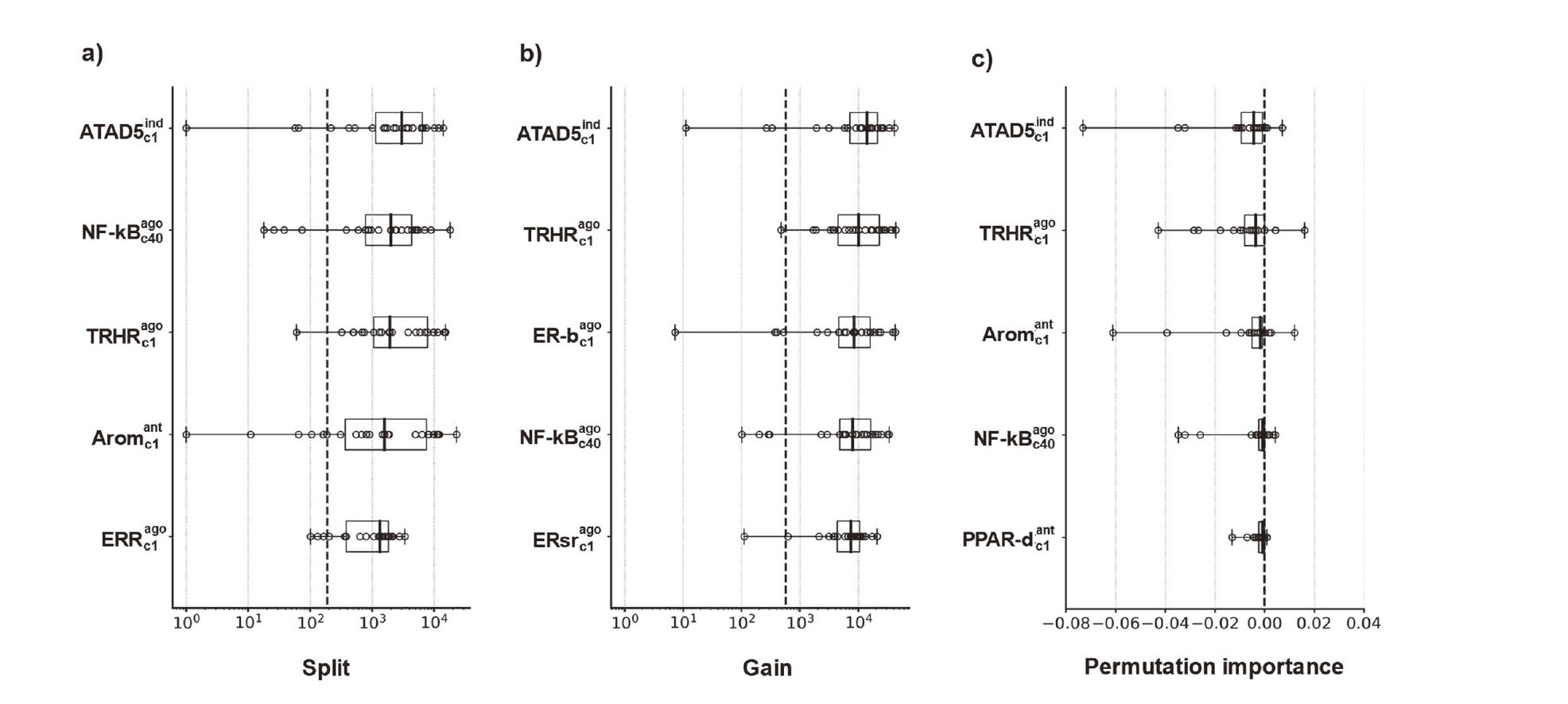
To evaluate the contribution of MIEs to LMT, LGB was used to evaluate the importance of a feature based on ‘split’, which defines the number of times the tree nodes are split, ‘gain’, which measures the actual decrease in node impurity, and ‘permutation importance’, which measures the actual decrease in the evaluated ROC–AUC. Because we adopted a nested fivefold CV modeling scheme, we generated 25 models for one algorithm. Therefore, the feature importance was evaluated according to the median value of 25 models. In the analysis of ‘split’ and ‘gain’, cases, a feature with zero importance were excluded from the analysis.
Evaluation of single MIE
Eighty-seven MIEs that decreased their average permutation importance based on ROC-AUC were included in this analysis. Logistic regression models (implemented by scikit-learn library) were constructed for each MIE to evaluate the predictive ability of that MIE alone. In this modeling process, only ensemble averaging in the inner fivefold CV was adopted (hyperparameter optimization was excluded). The predictive performance of each MIE was evaluated by ROC–AUC evaluation on the test sets (n = 5). The positive or negative contribution of each MIE activity to the prediction was evaluated from the regression coefficients of the logistic regression models (n = 25).
RESULTS
Predictive performance
The predictive performances of the five models were evaluated according to the ROC–AUC and MCC (Fig. 2). All models performed better under normal conditions than with y-randomization, particularly LGB and XGB. This confirmed that the performances were not due to chance correlation. ROC–AUC was independent of the cutoff point and thus was the most appropriate metric for separating the model performances. LGB provided the best generalizability and ROC–AUC (0.703 ± 0.054, n = 5) (Table 1).
Table 1. Predictive performances in the test sets.
 Contributing MIEs
Contributing MIEs
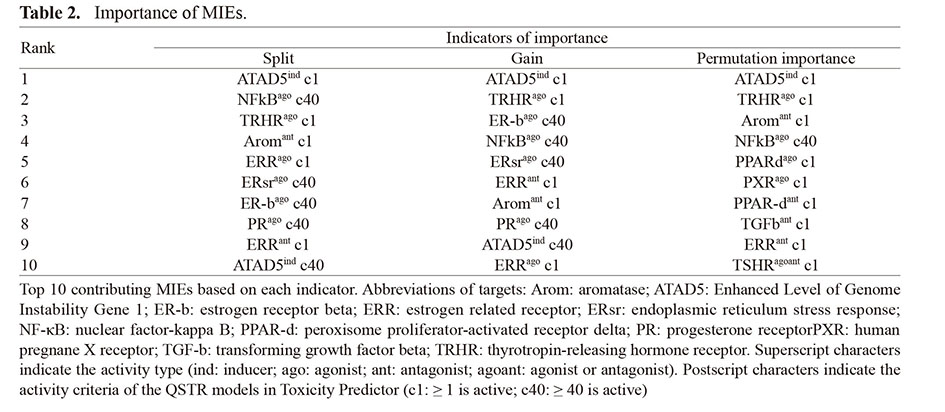
LGB was determined to have the best performance among the models, so it was applied to identifying important features that could contribute to predicting LMT-inducing drugs. For all indicators, the five most important features all had a higher importance than the median for all features (Fig. 3). The most important feature common to all indicators was the enhanced level of genome instability gene 1 (ELG1; human ATAD5). Activity of the thyrotropin-releasing hormone receptor (TRHR) agonist also showed a remarkable contribution to the model construction. The other important features included the agonist activity of nuclear factor-κ B (NF-κB). Table 2 shows the MIEs of the top 10 contributors to the prediction based on each importance indicator. The raw data of feature importance for all models and indicators is available in the Supplemental Information (SI. 3).
Table 2. Importance of MIEs.
 Prediction with single MIE
Prediction with single MIE
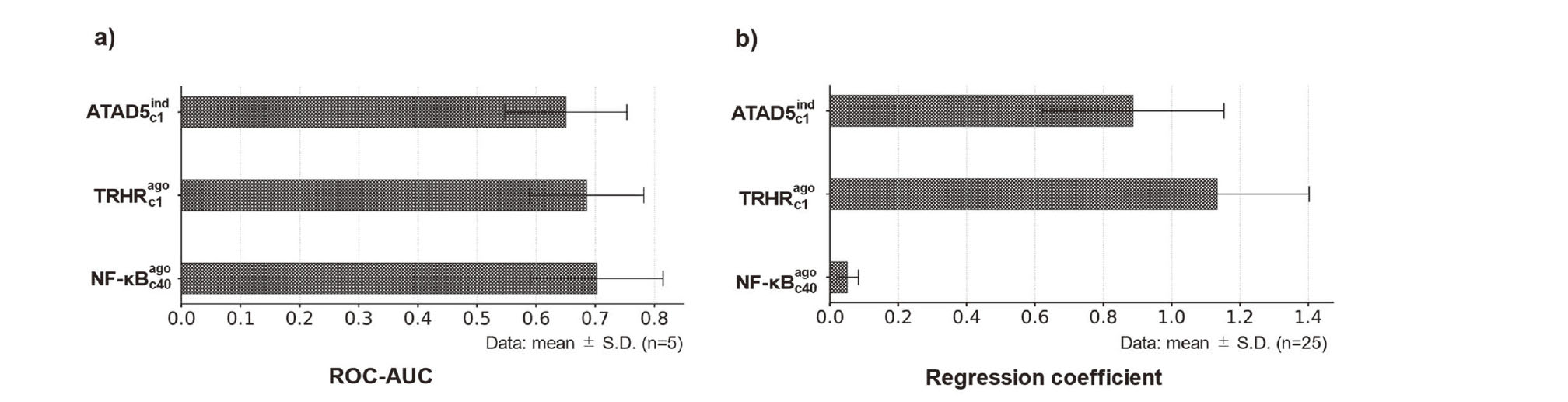
The MIEs with strong contributions common to all indicators were ATAD5 genotoxic inducer (criterion 1), TRHR agonist (criterion 1), and NF-κB (criterion 40) agonist activity. Each of these MIEs individually predicted the ROC–AUC in the test sets (ATAD5 genotoxic inducer: 0.650 ± 0.126; TRHR agonist: 0.686 ± 0.118; NF-κB agonist: 0.703 ± 0.137), indicating that they are remarkable predictors of LMT-inducing drugs (Fig. 4a; see also the Supplemental Information: Fig. S1). Moreover, the regression coefficients of all logistic regression models for these MIEs were positive (Fig. 4b and Supplemental Information: Fig. S1), emphasizing that the drugs with high predictive activity of ATAD5, TRHR, and NF-κB are LMT-inducing drugs.
Predicted LMT-inducing drug
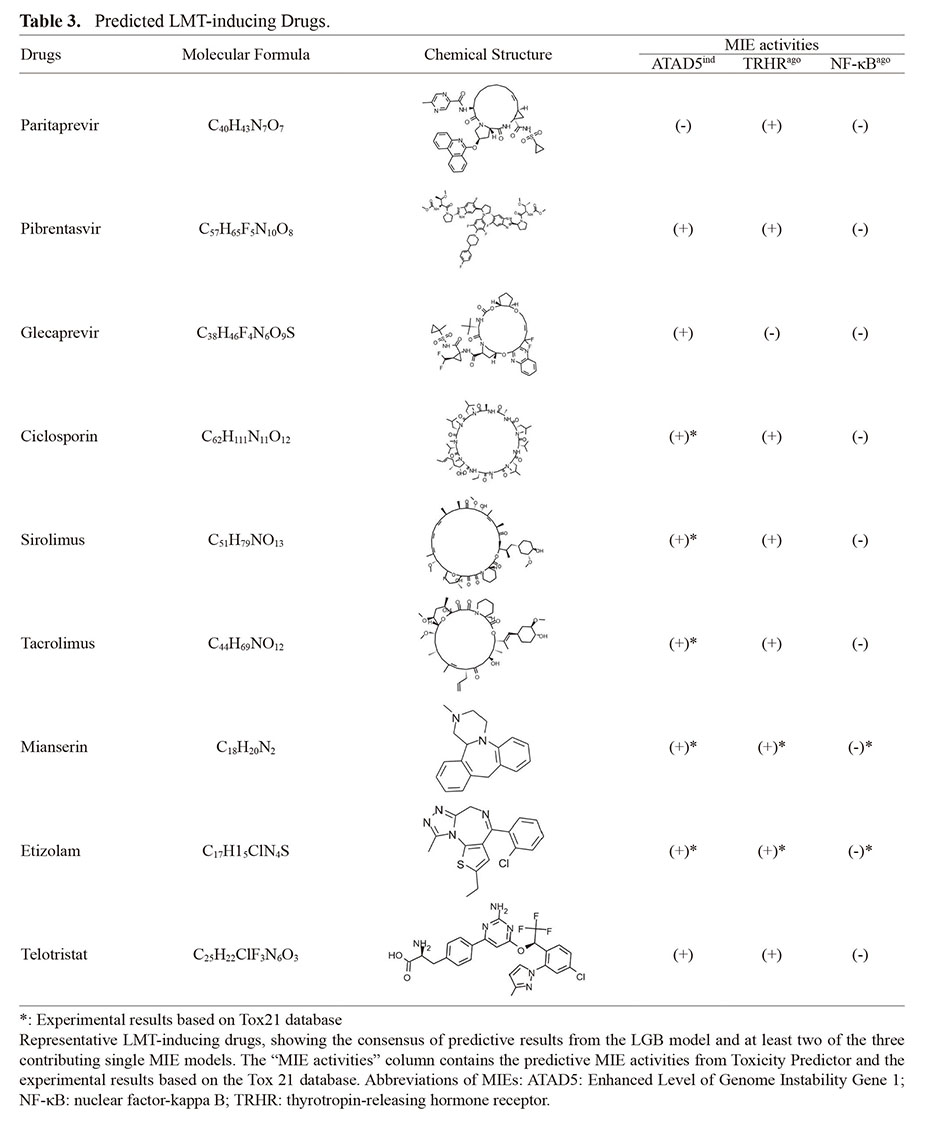
We detected nine representative drugs (paritaprevir, pibrentasvir, glecaprevir, ciclosporin, sirolimus, tacrolimus, mianserin, etizolam, and telotristat) with positive predictions by the LGB model and at least two single MIE models among ATAD5 genotoxic inducer (criterion 1), TRHR agonist (criterion 1), and NF-κB (criterion 40) (Table 3). These nine drugs were LMT-positive in the annotated dataset used in this study. Especially, positives of ATAD5 genotoxic inducer activity and TRHR agonist activity, predicted with Toxicity Predictor and curated from the Tox21 database, have been observed for many of nine drugs.
Table 3. Predicted LMT-inducing Drugs.
DISCUSSION
Recent studies have proposed hepatocarcinogens prediction models mediated by toxicogenomic data such as Open TG-GATEs (Huang and Tung, 2017; Yamada et al., 2016). In this study, using a drug-induced LMT dataset in humans annotated with the FAERS database and the MIE activities curated and predicted with Toxicity Predictor as the biological intermediate variables, we predicted LMT-inducing drugs in humans and offered their biological interpretation. Toxicity Predictor is based on the Tox21 database, the largest database in the U.S., which contains the chemical structures and toxicity assay results of more than 10K compounds, and therefore contains a wider range of compounds that can be predicted and complemented than compound-gene expression interaction databases such as Open TG-GATEs. In addition, ADEs such as hepatocarcinogenesis, which are difficult to assess by prospective studies in human populations, can be directly annotated based on ADEs in human populations in backward-looking studies on FEARS data.
Contributing MIEs to the prediction of LMT-inducing drug
In this study, we found that the activities of ATAD5, TRHR and NF-κB are valid predictors of LMT-inducing drugs. ATAD5 has been shown to increase in response to DNA damage (Fox et al., 2012) and thus has been used for identifying genotoxic compounds. ATAD5 is significantly higher in human cancers compared to normal liver cells (Ghosh et al., 2016). Our results suggest that ATAD5 is not only a biomarker of spontaneous hepatocellular carcinoma (HCC) but also that it can be used for ADE prediction of drug-induced LMT. Meanwhile, TRHR is a regulator of thyroid hormones, which work closely with the liver for cell proliferation, body weight regulation, lipogenesis, lipid metabolism, and insulin resistance. Modulation of these functions may affect the development of several liver diseases such as nonalcoholic fatty liver disease (NAFLD) (Gionfra et al., 2019). Recent epidemiological evidence has suggested that NAFLD such as hepatic steatosis is a major contributor to HCC (Anstee et al., 2019). Our prediction model supports the mechanism of NAFLD-related HCC and indicates that it can be predicted according to TRHR activity. NF-κB mediates both pro-inflammatory and anti-apoptotic responses to mediators such as cytokines. Experiments with mouse models have shown that genetic ablation of NF-κB regulators leads to spontaneous liver injury, fibrosis, and HCC (Luedde et al., 2007; Bettermann et al., 2010; Inokuchi et al., 2010). Too little or too much NF-κB activation may negatively affect the liver owing to increased inflammation or insufficient protection from cell death (Luedde and Schwabe, 2011). These findings emphasized that known MIEs related to carcinogenicity can be used for predicting LMT-inducing drugs annotated based on clinical reports.
On the other hand, although estrogenic functions were not consistently important, diverse modulatory effects of estrogenic function through estrogen receptors (ERs) and aromatase were also found to be remarkable as predictors of LMT-inducing drugs. The relevance of estrogen and ERs functions to hepatocarcinogenesis is controversial. While epidemiological evidence has been reported that estrogen function is protective of HCC because HCC incidence increases after menopause (Hassan et al., 2017), activation of estrogen-related receptors that are not activated by endogenous estrogen has been associated with tumorigenesis (Ranhotra, 2015). Our findings emphasized that the diverse modulatory effects of drugs on the estrogen function through ERs and aromatase indicate their significance as predictors of LMT-inducing drugs.
Predicted LMT-inducing drug
Anti-hepatitis C virus agents, such as paritaprevir, pibrentasvir and glecaprevir, and immunosuppressants, such as tacrolimus, sirolimus, and ciclosporin, which were predicted positive for drug-induced LMT, have MIE activities that contribute to drug-induced LMT prediction. These drugs are reportedly associated with hepatocarcinogenicity (Kogiso et al., 2018; Laprise et al., 2019). Especially, cyclosporine use has been associated with carcinogenesis of the lip, and cyclosporine is suspected to cause DNA damage and cell proliferation (Herman et al., 2001). Similarly, we found that hepatocarcinogenesis is associated with elevated levels of both ATAD5, which is involved in genomic instability, and TRHR activity, which is also associated with proliferative effects. These results confirm the validity of the LMT-inducing drugs predicted by our MIE-based prediction model.
Limitations
One limitation of this study is the limited proportion of LMT to DILI, because only 44 LMT-inducing drugs were identified, compared to the 197 most DILI-concerning drugs in DILIrank dataset. Therefore, as the LMT-induced drugs represent approximately at most 20% of severe DILI drugs, our model makes a limited contribution to the prior prediction of development discontinuation of DILI drugs, which is important from a drug-discovery perspective.
In conclusion, our results show that MIE activities are useful predictors of LMT-inducing drugs. We constructed prediction models to identify the significant features. ATAD5, TRHR, and NF-κB activities were identified as the most remarkable biological predictors of drug-induced LMT. This study is the first successful application of a post-marketing database in the prediction of drug-induced LMT in humans based on MIE activities. That is, drug-induced LMT can be predicted from the interaction between molecular exposure and a biosystem. The results of this study may provide useful knowledge for drug development, research, and regulatory decision-making and will contribute to the construction of more accurate and meaningful DILI prediction models by increasing our understanding of biologically interpretable predictors.
Conflict of interest
The authors declare that there is no conflict of interest.
REFERENCES
- Aguirre-Plans, J., Piñero, J., Souza, T., Callegaro, G., Kunnen, S.J., Sanz, F., Fernandez-Fuentes, N., Furlong, L.I., Guney, E. and Oliva, B. (2021): An ensemble learning approach for modeling the systems biology of drug-induced injury. Biol. Direct, 16, 5.
- Amano, Y., Honda, H., Sawada, R., Nukada, Y., Yamane, M., Ikeda, N., Morita, O. and Yamanishi, Y. (2020): In silico systems for predicting chemical-induced side effects using known and potential chemical protein interactions, enabling mechanism estimation. J. Toxicol. Sci., 45, 137-149.
- Anstee, Q.M., Reeves, H.L., Kotsiliti, E., Govaere, O. and Heikenwalder, M. (2019): From NASH to HCC: current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol., 16, 411-428.
- Bergstra, J., Yamins, D. and Learning, D.C. (2013): Making a science of model search: hyperparameter optimization in hundreds of dimensions for vision architectures. PMLR, 13, 115-123.
- Bettermann, K., Vucur, M., Haybaeck, J., Koppe, C., Janssen, J., Heymann, F., Weber, A., Weiskirchen, R., Liedtke, C., Gassler, N., Müller, M., de Vos, R., Wolf, M.J., Boege, Y., Seleznik, G.M., Zeller, N., Erny, D., Fuchs, T., Zoller, S., Cairo, S., Buendia, M.A., Prinz, M., Akira, S., Tacke, F., Heikenwalder, M., Trautwein, C. and Luedde, T. (2010): TAK1 suppresses a NEMO-dependent but NF-kappaB-independent pathway to liver cancer. Cancer Cell, 17, 481-496.
- Cawley, G.C. and Talbot, N.L. (2010): On over-fitting in model selection and subsequent selection bias in performance evaluation. J. Mach. Learn. Res., 11, 2079-2107.
- Chen, M., Suzuki, A., Thakkar, S., Yu, K., Hu, C. and Tong, W. (2016): DILIrank: the largest reference drug list ranked by the risk for developing drug-induced liver injury in humans. Drug Discov. Today, 21, 648-653.
- Diamanti-Kandarakis, E., Bourguignon, J.P., Giudice, L.C., Hauser, R., Prins, G.S., Soto, A.M., Zoeller, R.T. and Gore, A.C. (2009): Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr. Rev., 30, 293-342.
- Fawcett, T. (2006): An Introduction to ROC Analysis. Pattern Recognit. Lett., 27, 861-874.
- Fox, J.T., Sakamuru, S., Huang, R., Teneva, N., Simmons, S.O., Xia, M., Tice, R.R., Austin, C.P. and Myung, K. (2012): High-throughput genotoxicity assay identifies antioxidants as inducers of DNA damage response and cell death. Proc. Natl. Acad. Sci. USA, 109, 5423-5428.
- Gadaleta, D., Manganelli, S., Roncaglioni, A., Toma, C., Benfenati, E. and Mombelli, E. (2018): QSAR modeling of ToxCast assays relevant to the molecular initiating events of AOPs leading to hepatic steatosis. J. Chem. Inf. Model., 58, 1501-1517.
- Ghosh, A., Ghosh, S., Dasgupta, D., Ghosh, A., Datta, S., Sikdar, N., Datta, S., Chowdhury, A. and Banerjee, S. (2016): Hepatitis B virus X protein upregulates hELG1/ ATAD5 expression through E2F1 in hepatocellular carcinoma. Int. J. Biol. Sci., 12, 30-41.
- Gionfra, F., De Vito, P., Pallottini, V., Lin, H.Y., Davis, P.J., Pedersen, J.Z. and Incerpi, S. (2019): The role of thyroid hormones in hepatocyte proliferation and liver cancer. Front. Endocrinol. (Lausanne), 10, 532.
- Hardt, A., Stippel, D., Odenthal, M., Hölscher, A.H., Dienes, H.P. and Drebber, U. (2012): Development of hepatocellular carcinoma associated with anabolic androgenic steroid abuse in a young bodybuilder: a case report. Case Rep. Pathol., 2012, 195607.
- Hassan, M.M., Botrus, G., Abdel-Wahab, R., Wolff, R.A., Li, D., Tweardy, D., Phan, A.T., Hawk, E., Javle, M., Lee, J.S., Torres, H.A., Rashid, A., Lenzi, R., Hassabo, H.M., Abaza, Y., Shalaby, A.S., Lacin, S., Morris, J., Patt, Y.Z., Amos, C.I., Khaderi, S.A., Goss, J.A., Jalal, P.K. and Kaseb, A.O. (2017): Estrogen replacement reduces risk and increases survival times of women with hepatocellular carcinoma. Clin. Gastroenterol. Hepatol., 15, 1791-1799.
- He, S., Ye, T., Wang, R., Zhang, C., Zhang, X., Sun, G. and Sun, X. (2019): An in silico model for predicting drug-induced hepatotoxicity. Int. J. Mol. Sci., 20, 1897.
- Herman, M., Weinstein, T., Korzets, A., Chagnac, A., Ori, Y., Zevin, D., Malachi, T. and Gafter, U. (2001): Effect of cyclosporin A on DNA repair and cancer incidence in kidney transplant recipients. J. Lab. Clin. Med., 137, 14-20.
- Hong, H., Thakkar, S., Chen, M. and Tong, W. (2017): Development of decision forest models for prediction of drug-induced liver injury in humans using a large set of FDA-approved drugs. Sci. Rep., 7, 17311.
- Huang, S.H. and Tung, C.W. (2017): Identification of consensus biomarkers for predicting non-genotoxic hepatocarcinogens., 7, 41176.
- Inokuchi, S., Aoyama, T., Miura, K., Osterreicher, C.H., Kodama, Y., Miyai, K., Akira, S., Brenner, D.A. and Seki, E. (2010): Disruption of TAK1 in hepatocytes causes hepatic injury, inflammation, fibrosis, and carcinogenesis. Proc. Natl. Acad. Sci. USA, 107, 844-849.
- Kogiso, T., Sagawa, T., Kodama, K., Taniai, M., Katagiri, S., Egawa, H., Yamamoto, M. and Tokushige, K. (2018): Hepatocellular carcinoma after direct-acting antiviral drug treatment in patients with hepatitis C virus. JGH Open, 3, 52-60.
- Kurosaki, K., Wu, R. and Uesawa, Y. (2020): A toxicity prediction tool for potential agonist/antagonist activities in molecular initiating events based on chemical structures. Int. J. Mol. Sci., 21, 7853.
- Kurosaki, K. and Uesawa, Y. (2021): Molecular initiating events associated with drug-induced liver malignant tumors: an integrated study of the FDA adverse event reporting system and toxicity predictions. Biomolecules, 11, 944.
- Laprise, C., Cahoon, E.K., Lynch, C.F., Kahn, A.R., Copeland, G., Gonsalves, L., Madeleine, M.M., Pfeiffer, R.M. and Engels, E.A. (2019): Risk of lip cancer after solid organ transplantation in the United States. Am. J. Transplant., 19, 227-237.
- Liu, Z., Zhu, L., Thakkar, S., Roberts, R. and Tong, W. (2020): Can Transcriptomic Profiles from Cancer Cell Lines Be Used for Toxicity Assessment? Chem. Res. Toxicol., 33, 271-280.
- Luedde, T. and Schwabe, R.F. (2011): NF-κB in the liver--linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol., 8, 108-118.
- Luedde, T., Beraza, N., Kotsikoris, V., van Loo, G., Nenci, A., De Vos, R., Roskams, T., Trautwein, C. and Pasparakis, M. (2007): Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell, 11, 119-132.
- Matthews, B.W. (1975): Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta, 405, 442-451.
- Mizushima, T. and Miyamoto, H. (2019): The Role of Androgen Receptor Signaling in Ovarian Cancer. Cells, 8, 176.
- Namani, A., Li, Y., Wang, X.J. and Tang, X. (2014): Modulation of NRF2 signaling pathway by nuclear receptors: implications for cancer. Biochim. Biophys. Acta, 1843, 1875-1885.
- Onakpoya, I.J., Heneghan, C.J. and Aronson, J.K. (2016): Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: a systematic review of the world literature. BMC Med., 14, 10.
- Ranhotra, H.S. (2015): Estrogen-related receptor alpha and cancer: axis of evil. J. Recept. Signal Transduct. Res., 35, 505-508.
- Safe, S., Lee, S.O. and Jin, U.H. (2013): Role of the aryl hydrocarbon receptor in carcinogenesis and potential as a drug target. Toxicol. Sci., 135, 1-16.
- Solbach, P., Potthoff, A., Raatschen, H.J., Soudah, B., Lehmann, U., Schneider, A., Gebel, M.J., Manns, M.P. and Vogel, A. (2015): Testosterone-receptor positive hepatocellular carcinoma in a 29-year old bodybuilder with a history of anabolic androgenic steroid abuse: a case report. BMC Gastroenterol., 15, 60.
- Stoot, J.H., Coelen, R.J., De Jong, M.C. and Dejong, C.H. (2010): Malignant transformation of hepatocellular adenomas into hepatocellular carcinomas: a systematic review including more than 1600 adenoma cases. HPB (Oxford), 12, 509-522.
- Wang, Y., Xiao, Q., Chen, P. and Wang, B. (2019): In silico prediction of drug-induced liver injury based on ensemble classifier method. Int. J. Mol. Sci., 20, 4106.
- Woodward, C., Smith, J., Acreman, D. and Kumar, N. (2019): Hepatocellular carcinoma in body builders; an emerging rare but serious complication of androgenic anabolic steroid use. Ann. Hepatobiliary Pancreat. Surg., 23, 174-177.
- Yamada, F., Sumida, K. and Saito, K. (2016): An improved model of predicting hepatocarcinogenic potential in rats by using gene expression data. J. Appl. Toxicol., 36, 296-308.
- Zhu, X. and Kruhlak, N.L. (2014): Construction and analysis of a human hepatotoxicity database suitable for QSAR modeling using post-market safety data. Toxicology, 321, 62-72.
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/47_89-t1.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/47_89-t2.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/47_89-t3.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)