INTRODUCTION
During drug discovery and development stage, prediction of metabolite profiles in humans is important to elucidate the pharmacological and toxicological potential of drug candidates. In 2008, the US Food and Drug Administration (FDA) issued a guidance on “Metabolites in Safety Testing” (MIST). In addition, the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) issued the “M3(R2) Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals” guideline, which recommends that evaluation of metabolites is warranted when the metabolites are observed at exposures > 10% of total drug-related material exposure and at significantly greater levels in humans than the maximum exposure seen in the toxicity studies. Therefore, evaluation of drug metabolites during drug discovery and development stage has become important.
Although in vitro systems using human materials are employed to predict human metabolites, Dalvie et al. (2009) reported that the accuracy of prediction for Phase I metabolites was only 50% to 69% and that for secondary metabolites was < 47% in primary human hepatocytes. These poor prediction results might be due to the function of enzymes involved in metabolism being affected by the preparation, storage, and experimental treatment of hepatocytes (Wang et al., 2005). To the best of our knowledge, the accurate prediction of human metabolites by in vitro systems remains to be difficult.
As in vivo systems to predict human metabolites, several humanized animal models have been developed recently. Some of these models insert and express human drug metabolism-related genes such as cytochrome P450 (CYP) 1A1/2, CYP2D6, CYP2E1, and CYP3A4 (Muruganandan and Sinal, 2008) and the CYP3A cluster (Kazuki et al., 2013). An alternate approach involves producing chimeric mice with humanized liver by transplanting human hepatocytes into liver-injured immunodeficient mice that can accept xenografted human hepatocytes in their liver. Several different methods for inducing liver injury and different immunodeficiencies have been investigated (Peltz, 2013), and chimeric mice with humanized liver generated using urokinase-type plasminogen activator/severe combined immunodeficiency (uPA/SCID) mice repopulated with human hepatocytes (PXB-mice; PhoenixBio Co., Ltd., Hiroshima, Japan) have been reported (Tateno et al., 2015). These mice are repopulated with approximately 80% of human hepatocytes. Expression levels and activities of P450s and non-P450 enzymes, such as uridine 5’-diphospho-glucuronosyl transferase (UGT) and sulfotransferase (SULT), in the liver of PXB-mice are similar to those in humans (Katoh et al., 2004, 2005; Katoh and Yokoi, 2007; Kitamura et al., 2008; Nishimura et al., 2005; Ohtsuki et al., 2014), and human-specific metabolites are also formed in PXB-mice (De Serres et al., 2011; Inoue et al., 2009; Kamimura et al., 2010; Yamazaki et al., 2010). In addition, Kakuni et al. (2012) reported the production of troglitazone-induced liver injury in PXB-mice, suggesting that chimeric mice with humanized liver are a new animal model for studying idiosyncratic drug-induced liver injury.
Felbamate (FBM) is an antiepileptic drug used for the treatment of partial and generalized seizures, including Lennox-Gastaut syndrome in children (O’Neil et al., 1996). FBM was approved by the Food and Drug Administration (FDA) in 1993. After the launch of FBM, more than 100,000 patients were treated with FBM, and more than 50 events of aplastic anemia or hepatotoxicity were reported as FBM-related adverse effects (Kaufman et al., 1997). FBM-induced liver and bone marrow toxicities were not observed in the phase III clinical trial. Therefore, the toxicities are thought to be idiosyncratic drug toxicity (IDT). A previous study provided the evidence that reactive metabolites of drugs are responsible for IDT (Park et al., 1998), and it has been reported that the reactive metabolite responsible for FBM-induced IDT is α, β-unsaturated metabolite, 2-phenylpropenal (2-PP) (Thompson et al., 1996). FBM is initially oxidized to p-hydroxy FBM (pOH-FBM), 2-hydroxy FBM (2OH-FBM) or hydrolyzed to monocarbamate felbamate (MCF). MCF undergoes oxidation to aldehyde felbamate (CBMA), which spontaneously forms 2-PP. The postulated metabolic pathways of FBM to 2-PP, as well as other metabolites, are shown in Fig. 1 (Dieckhaus et al., 2001, 2002; Kapetanovic et al., 2002).

Regarding the metabolism of FBM, a chemical inhibition study using CYP inhibitors and liver microsomes suggested the involvement of several CYP isoforms (Glue et al., 1997); however, to the best of our knowledge, no studies have been reported for the hydrolyzing enzymes involved in the metabolism of FBM to MCF. In this study, we used human CYP and carboxylesterase (CES) expressing microsomes to further investigate the enzymes involved in the metabolism of FBM and the susceptibility that contribute to IDT of FBM. In addition, we investigated the metabolite profiles of FBM in PXB-mice, which possess human hepatocytes, to clarify the species difference of IDT. Furthermore, we evaluated the change in distribution of glutathione (GSH), which has crucial role in the detoxification of reactive metabolite in PXB-mice liver after administration of FBM by mass spectrometry imaging (MSI) analysis. MSI can evaluate the change in distribution of not only drug substances but also endogenous molecules. Previous studies reported that distribution of drugs and their metabolites in organs have been visualized by MSI (Sugiura and Setou, 2010; Takai and Tanaka, 2015). Changes of endogenous molecules between normal and tumor tissues (Goto et al., 2014; Kawashima et al., 2013; Shimma et al., 2007), and changes in response to drugs were also examined using MSI (Sun et al., 2016). Based on the obtained data, we have made a comprehensive consideration of FBM metabolism and susceptibility to drug-induced idiosyncratic hepatotoxicity.
MATERIALS AND METHODS
Chemicals and reagents
Felbamate (FBM) was purchased from Sigma-Aldrich Laboratories (St. Louis, MO, USA) and AK Scientific, Inc. (Union City, CA, USA). 2,5-dihydroxybenzoic acid (DHB) was purchased from Tokyo Chemical Industry (Tokyo, Japan). NADPH-regenerating system regents (BD Biosciences, Woburn, MA, USA) were used as the reaction mixture for the in vitro metabolism study. All other reagents used were commercially available and of guaranteed purity.
Equipment
The following systems were used to conduct liquid chromatography-mass spectrometry (LC-MS): Agilent 1290 Infinity (Agilent Technologies, Inc., Palo Alto, CA, USA) coupled with Orbitrap Velos Pro (Thermo Fisher Scientific, Waltham, MA, USA). Solarix XR (Bruker Daltonics K.K., Bremen, Germany) was used for mass spectrometry imaging of liver tissue slices.
Human CES and CYP expressing microsomes
Human CYP1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 2J2, 3A4, 3A5, and CES1b, CES1c, CES2 expressing microsomes, and control microsomes, which do not contain specific CYP and CES isoforms were purchased from CORNING (Corning, NY, USA) and stored at −80°C until use.
Animals
Chimeric mice with humanized liver (PXB-mice®, PhoenixBio Co., Ltd., Hiroshima, Japan) were prepared by transplantation of commercially available human hepatocytes (BD Biosciences, Woburn, MA, USA) into cDNA-urokinase-type plasminogen activator-transgenic severe combined immunodeficient mice (cDNA-uPAwild/+/SCID, Tateno et al., 2015). The animal study was approved by the Animal Care and Use Committee of PhoenixBio Co., Ltd., and Hiroshima University.
In vitro metabolic studies using human CES and CYP expressing microsomes
Human CYP and CES expressing microsomes (1 mg protein/mL for CES and 100 pmol/mL for CYP) were incubated in phosphate buffered saline (pH 7.4) in the presence of FBM (10 µmol/L) at 37°C. After incubation for 120 min, metabolic reaction was stopped by addition of ice-cold acetonitrile and centrifuged at 3,000 rpm for 10 min at 4°C. The supernatant was filtered via centrifugal filtration, and the filtrate was subjected to LC-MS. Mobile phases consisted of water/formic acid (1000:1, Solvent A) and acetonitrile/formic acid (1000:1, Solvent B) on ACQUITY UPLC BEH 18 (1.7 μm, 2.1 × 100 mm; Waters) at 40°C. Chromatographic separation was conducted in gradient elution. The proportion of solvent B was maintained 5% from 0 to 3 min and then linearly increased to 70% from 3 to 12 min and maintained at 70% to 15 min. The remaining amount of FBM was monitored by LC-MS using accurate mass of FBM at positive ion mode m/z = 239.10140
In vivo metabolite profiling using PXB-mice
FBM in 0.5% carboxymethylcellulose was orally administered at 600 mg/kg to PXB-mice (n = 3). Blood samples were collected 2 hr after administration and centrifuged to obtain plasma. Liver samples were collected 2 hr after administration and mixed with phosphate buffered saline and homogenated to obtain 20% liver homogenate. A 100-μL aliquot of plasma and liver homogenate samples (n = 3, each) were mixed to obtain the pooled plasma and liver homogenate samples (n = 1, each). The pooled samples were mixed with equivalent volume of acetonitrile and centrifuged at 3,000 rpm for 10 min at 4°C. The supernatant was filtered via centrifugal filtration, and the filtrate was subjected to LC-MS. Mobile phases consisted of water/formic acid (1000:1, Solvent A) and acetonitrile/formic acid (1000:1, Solvent B) on a CAPCELL PAK C18 AQ (3 μm, 3 × 250 mm; Shiseido) at 40°C. Chromatographic separation was conducted in gradient elution. The proportion of solvent B was maintained at 0% from 0 to 10 min and then linearly increased to 95% from 10 to 95 min and maintained at 100% from 95 to 100 min. Sample eluents were monitored by UV absorbance at 210 nm and MS at m/z 100–1000. Metabolites were estimated by the retention times, molecular weights, and the fragment patterns of the metabolites.
Mass spectrometry imaging in PXB-mice liver
PXB-mice livers were collected 24 hr after administration of FBM (600 mg/kg) in 0.5% methylcellulose, and the collected liver samples were frozen on dry ice. Liver sections (10 μm) were prepared with cryomicrotome (CRYO STAR NX70, Thermo Fisher Scientific) and mounted on glass slides (Matsunami Glass Co., Ltd., Osaka, Japan). DHB (10 mg/mL in 50% methanol containing 0.1% TFA) was applied onto the glass slide and analyzed by Solarix XR. A laser scan was carried out on the liver slice automatically using MSI software ftms control (Bruker Daltonics K.K). MS spectra were acquired in positive ion mode at a frequency of 1000 Hz within the m/z range from 100–600. Changes in ion intensity [M + K]+ corresponding to FBM (m/z = 277.0585), and endogenous biomolecule GSH (m/z = 346.0470) were analyzed using MSI software flexImaging (Bruker Daltonics K.K).
Blood biochemical test
Plasma alanine aminotransferase (ALT), aspartate aminotransferase (AST) and alkaline phosphatase (ALP) levels at 24 hr after administration of FBM (600 mg/kg) were measured using FUJI DRI-CHEM analyzer (FUJIFILM, Tokyo, Japan).
Statistical analysis
The statistical analysis was performed using the t-test and Dunnett’s test. A p-value < 0.05 was considered significant.
RESULTS AND DISCUSSION
Identification of human liver enzymes involved in the metabolism of FBM
FBM is reported to be initially oxidized to pOH-FBM, 2OH-FBM or hydrolyzed to MCF, and reactive metabolite, 2-PP, is produced through the formation of MCF. It is important to reveal the metabolic enzymes involved in the oxidation and hydrolyzation pathways because oxidation is related to avoidance of 2-PP formation and hydrolyzation is related to the formation of 2-PP. Previous studies showed that P450 enzymes were involved in the formation of pOH-FBM and 2OH-FBM and esterase were involved in the formation of MCF (Dieckhaus et al., 2002; Glue et al., 1997). Therefore, in vitro CYP and CES reaction phenotyping systems were used to clarify the enzymes involved in the metabolism of FBM to pOH-FBM, 2OH-FBM, and MCF.
FBM (10 μmol/L) was incubated with human CYP1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 2J2, 3A4, 3A5 isozymes or human CES1b, CES1c, CES2 isozymes for 120 min and the remaining amount of FBM was determined by LC-MS. After incubation for 120 min, FBM was significantly decreased in incubation with CYP2C8, 2C9, 2E1 isozymes and CES1c isozymes (Fig. 2). These results suggest that CYP2C8, 2C9, 2E1 and CES1c isozymes were involved in the metabolism of FBM.

Glue et al. (1997) reported that CYP2E1 and 3A4 were involved in the metabolism of FBM using human liver microsomes and chemical inhibitors for CYP isozymes. In our study, FBM was significantly decreased in CYP2E1 expressing microsomes, but not in CYP3A4. The reason for the inconsistence is unclear at the moment; however, differences in experimental conditions, such as chemical inhibition of CYP enzymes or reaction phenotyping systems might affect the results. Martignoni et al. (2006) reported that there are species differences in CYP2C family, and more Cyp2c isoforms were expressed in rodents than in humans, and it is thought that Cyp2c family is the major metabolic enzymes in rodent. It is possible that CYP2C-mediated production of oxidative metabolites (2OH-FBM and pOH-FBM) was higher in rodents than in humans, and thus toxicity by reactive metabolite (2-PP) could not be detected in experimental animals.
As for CES enzymes, human CES1 is reported to be one of the most highly expressed drug metabolizing enzymes in the liver, while human intestine only expresses CES2, and CES enzymes have moderate to high inter-individual variability (Di, 2019). Rodent CES enzymes are reported to have high catalytic capacity and efficiency, and there are a large number of CES isoforms expressed in the rodent liver (Di, 2019). This study has not tested which CES isoforms are involved in FBM metabolism in rodents. Given that the sequence homologies of CES between humans and rodents is 67–77% (Taketani et al., 2007; Hosokawa, 2008), the substrate recognition of CES may differ between humans and animals.
In addition, it has been reported that CYP2C9 has a genetic polymorphism and that the metabolism of many drugs, such as tolbutamide, phenytoin, and warfarin, is affected by the genetic polymorphism (Goldstein, 2001).
Taken together, species difference in CYP2C isoforms, genetic polymorphism of CYP2C9 and species difference of CES might affect the oxidation to pOH-FBM, 2OH-FBM and reduction to MCF and subsequent reactive metabolite formation and susceptibility to IDT between experimental animals and humans.
In vivo metabolite profiling of FBM in PXB-mice
It was reported that more than 50% of radioactivity was excreted into urine after administration of 14C radiolabeled FBM ([14C]FBM) to experimental animals and humans (Shumaker et al., 1990); therefore, metabolite profiles in urine were thought to be useful to consider the species difference of FBM metabolisms. In addition, Dieckhaus et al. (2000) reported that the amount of Nac-OH plus Nac-acid, which are N-acetylcysteine conjugated metabolites generated after GSH conjugation of 2-PP, was 6.3% and pOH-FBM plus 2OH-FBM was 10% of dose in human urine, whereas the amount of Nac-OH plus Nac-acid was lower (1.1%), and pOH-FBM plus 2OH-FBM was higher (62%) in rat urine. In addition, FBM was metabolized by CYP2C8 expressing microsomes in our study. These findings suggest that the amount of the reactive metabolite 2-PP might be different between experimental animals and human, and the difference might take part in IDT of FBM.
To investigate the formation of 2-PP in humans, a humanized animal model, PXB-mice, was used in this study. FBM was orally administered at 600 mg/kg to PXB-mice. FBM and its metabolites were analyzed in plasma and liver homogenate using LC-MS, and the chemical structures of metabolites were estimated using the results of MSn fragmentation. In UV chromatograms of the plasma samples, FBM, OH-FBM, CBMA, CCMF, MCF, and Nac-OH were detected. CPPA, MCF-gluc, MW441, and Nac-acid were also detected in plasma as trace level metabolites by MS detection (Fig. 3A). In the PXB-mouse liver samples, FBM was detected in UV chromatograms, and OH-FBM, CPPA, CBMA, CCMF, MCF, and MW441were also detected at trace levels (Fig. 3B). Various metabolites, including indicators of reactive metabolite formation (MW441, Nac-acid, Nac-OH), were detected in PXB-mice. Comparison of UV peak area of FBM and its metabolites are shown in Table 1.

Table 1. Comparison of UV peak area of FBM and its metabolites in plasma after administration of FBM at 600 mg/kg to PXB-mice.
|
CBMA
or
CCMF |
CBMA
or
CCMF |
2OH-FBM
or
pOH-FBM |
2OH-FBM
or
pOH-FBM |
MCF |
MW441 |
CPPA |
FBM |
MCF-gluc |
Nac-OH |
Nac-acid |
| UV Peak Area |
329052 |
161698 |
596233 |
1789046 |
1538782 |
Trace |
Trace |
118625182 |
Trace |
2115013 |
Trace |
| % of FBM |
0.28 |
0.14 |
0.50 |
1.51 |
1.30 |
- |
- |
100 |
- |
1.78 |
- |
Trace: only detected by MS.
It is important to evaluate drug metabolites in plasma from the viewpoint of regulations such as ICH and MIST guidance. In this study, many metabolites which were the indicators of reactive metabolite formation were detected in PXB-mice. Therefore, metabolite profiling using chimeric mice with humanized liver may be useful in assessing the risk of drug toxicity at the non-clinical stage. In addition, drug metabolism in human liver can be directly evaluated by using chimeric mouse liver homogenate. Reactive metabolites are generally known to be highly reactive, and it is thought that more detailed evaluation can be obtained by conducting studies on liver samples from chimeric mice.
Distribution of FBM and GSH in PXB-mouse liver
We investigated the metabolite profiles of FBM using PXB-mice, and various metabolites were detected in PXB-mouse liver. To elucidate the distribution of biological molecules after administration of FBM, MSI was performed using PXB-mouse liver sections. FBM was orally administered at 600 mg/kg to PXB-mice, and the liver tissue were collected at 24 hr after administration.
FBM was largely detected in PXB-mouse liver and distribution of the biological molecule GSH, associated with the detoxification of the reactive metabolite (2-PP) was also analyzed. In PXB-mouse liver, GSH tended to be decreased after administration of FBM, suggesting that 2-PP is removed by GSH conjugation (Fig. 4).
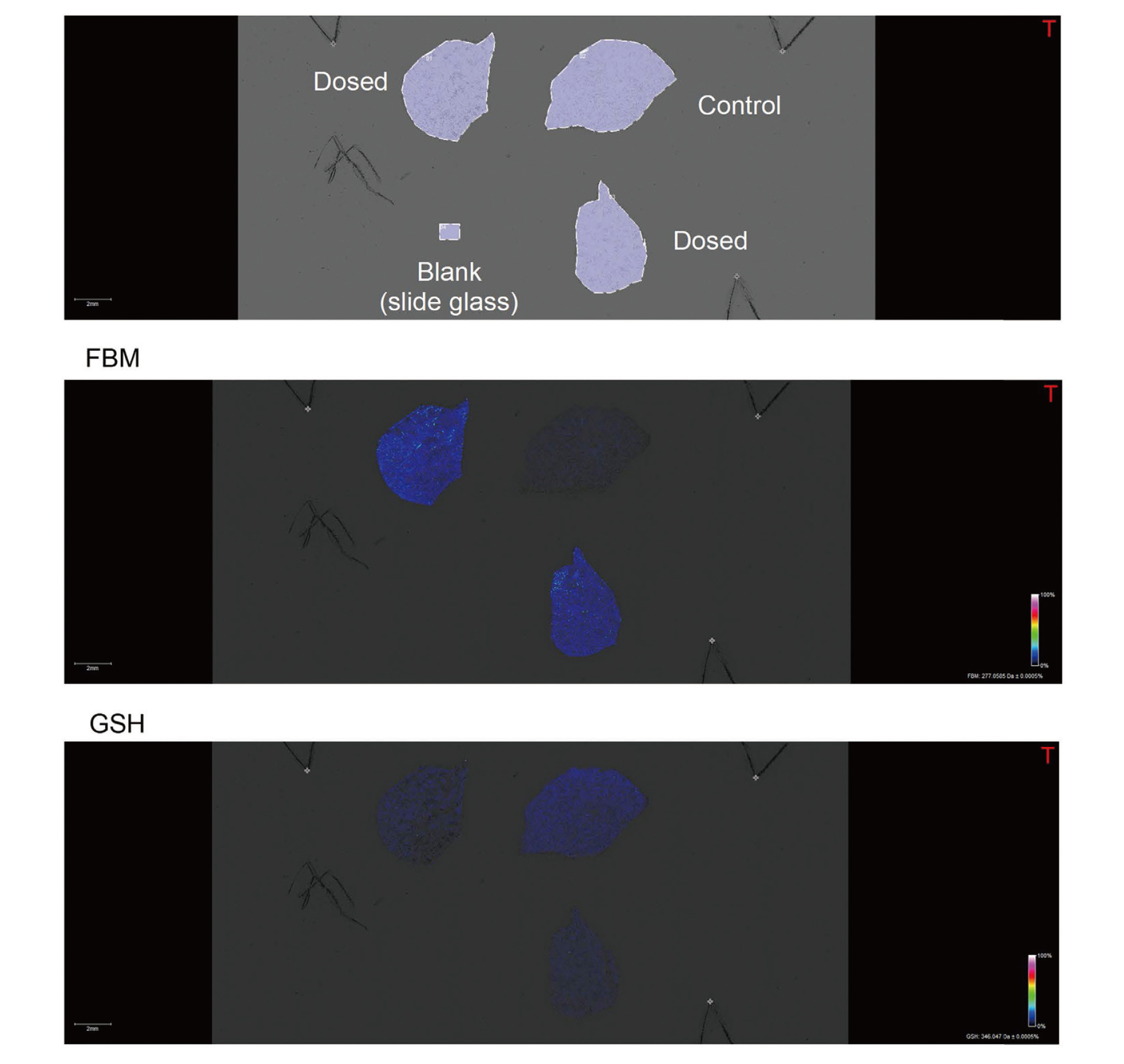
The reactive metabolite of FBM, 2-PP, has been reported to undergo GSH conjugation. Therefore, it seems reasonable that GSH levels in PXB-mouse liver were reduced after administration of FBM. By using chimeric mice and MSI analysis; it was possible to evaluate the effect of reactive metabolite, 2-PP, on human liver tissue in in vivo. The use of chimeric mice and MSI analysis are thought to be useful in investigating the liver toxicity of reactive metabolites in humans. In addition, Sanoh et al. (2017) reported the species difference in the changes of endogenous phospholipids caused by amiodarone administration using chimeric mice with middle replacement index. Since we used chimeric mice with high replacement index in this study, it is difficult to examine such species differences from a single field of view by MSI analysis, however, by using middle replacement chimeric mice, it may be possible to examine the species difference of drug metabolism and/or endogenous molecules from a single field of view by MSI analysis.
Blood biochemical test
Plasma ALT, AST and ALP levels were measured after administration of FBM at 600 mg/kg to PXB-mice. None of the parameters changed compared to the control group (Fig. 5). McGee et al. (1998) reported that no signs of toxicity were observed in mice after single oral administration of 3000 to 5000 mg/kg of FBM. In addition, after repeated oral administration of 300 to 600 mg/kg of FBM to rats and dogs, transient increase of ALT, AST and ALP occurred; however, no histological changes were observed in rat and dog liver (McGee et al., 1998).

In our study, it was considered that hepatotoxicity did not occur after administration of FBM because plasma ALT, AST and ALP levels of the FBM dosing group were comparable to those of the control group. These results are similar to a previous report using mice, rats and dogs (McGee et al., 1998). Taken together, further studies, such as repeated dose or ascending dose of FBM to PXB-mice, are required to clarify the relationship between reactive metabolite formation and hepatotoxicity of FBM. On the other hand, since chimeric mice with humanized liver have an immunodeficient background, it might be difficult to detect hepatotoxicity in PXB-mice if FBM-induced liver toxicity is immune-mediated. Therefore, it is also important to elucidate the detailed mechanism of IDT by FBM.
In conclusion, in this study, we revealed the metabolic enzymes involved in the metabolism of FBM to pOH-FBM, 2OH-FBM, and MCF. Although the effects of metabolism by residual mouse hepatocytes or small intestine might need to be further clarified, many metabolites after GSH conjugation of 2-PP were detected in chimeric mice with humanized liver. The GSH level in PXB-mouse liver tended to decrease after administration of FBM. Judging from these results, species difference of CYP and CES isozymes might affect the metabolism of FBM, and subsequently the formation of the reactive metabolite 2-PP in experimental animals and humans. Genetic polymorphism of CYP and CES isozymes might cause inter-individual difference in the metabolism of FBM, making it difficult to estimate the IDT of FBM. GSH level is thought to be, at least in part, related to liver toxicity in human. In addition, since chimeric mice with humanized liver well reflect the metabolism of FBM in humans, the humanized animal model is considered to be useful for elucidating the mechanism of IDT.