Abstract
The accumulation of excessively high manganese levels within the brain can contribute to a series of Parkinsonian symptoms referred to as manganism. The gasoline antiknock additive Methylcyclopentadienyl Manganese Tricarbonyl (MMT) is an environmental source of manganese exposure and can induce manganism in rats. While some prior reports have demonstrated the differential expression of small noncoding RNAs (sncRNAs) in patients with Parkinson’s disease (PD), the degree of sncRNA dysfunction in manganism has yet to be clearly documented. As sncRNAs such as transfer RNA-derived small RNAs (tsRNAs) and ribosomal RNA-derived small RNAs (rsRNAs) exhibit high levels of modifications such as 3’ terminal 3’-phosphate and 2’,3’-cyclic phosphate modifications that disrupt the process of adapter ligation and m1A, m3C, m1G, and m22G RNA methylation, these transcripts are not detected in traditional small RNA-sequencing studies. Here, differential sncRNA expression was analyzed by comparing a rat model of MMT-induced unrepaired striatum damage to appropriate control samples via PANDORA-Seq, which can detect highly modified sncRNAs. Following the removal of sncRNA modifications, this approach identified 599 sncRNAs that were differentially expressed in the striatum of MMT-exposed rats relative to controls, as well as 1155 sncRNAs that were differentially expressed in Mn-treated and control rats. Additional functional analyses were performed to predict the putative targets of these sncRNAs, implicating a role for such sncRNA dysregulation in the pathogenesis of manganism in this rat model system.
INTRODUCTION
The accumulation of excessively high concentrations of manganese (Mn) in the brain, particularly in the striatum, substantia nigra (SN), subthalamic nuclei, and globus pallodus, can contribute to the development of Parkinson’s disease (PD)-like symptoms referred to as manganism (Calne et al., 1994; Dorman et al., 2004; Kwakye et al., 2015). The first reports of manganism were published in 1837 following examinations of workers who were exposed in a factory responsible for the crushing of Mn-containing ore (Couper, 1837). Patients suffering from Mn toxicity primarily exhibit sensorimotor disorders, cognitive deficits, and abnormal neuropsychiatric function (Bakthavatsalam et al., 2014; Scholten, 1953). Dyskinesia in these patients often takes the form of specific motor impairments such as bradykinesia, hypertonia with cogwheel rigidity, “cock-walk” gait, a higher risk of falling when walking backward, and rapid postural tremors (Aschner and Aschner, 1991; Racette, 2014). Unlike PD, manganism is not associated with the formation of Lewy bodies, and affected patients are unresponsive to levodopa treatment. Individuals affected by Mn toxicity are also more likely to experience frequent dystonia than PD patients while exhibiting less severe resting tremors and failure of positron emission tomography (PET) studies to detect fluorodopa uptake (Kulshreshtha et al., 2021; Lu et al., 1994; Perl and Olanow, 2007; Wolters et al., 1989). Both PD and manganism are associated with widespread stiffness and bradykinesia, and both are driven by similar pathological mechanisms, including impaired proteasomal and autophagic activity, oxidative stress, aberrant signaling, cell death, mitochondrial dysfunction, and excitability-associated toxicity (Kwakye et al., 2015).
Environmental sources of Mn exposure include airborne Mn derived from the gasoline antiknock additive Methylcyclopentadienyl Manganese Tricarbonyl (MMT), the utilization of permanganate to purify drinking water, and occupational exposure to agrochemicals, including Maneb and Mancozeb (Röllin and Nogueira, 2011). Exposure to MMT or MnCl2 can promote the development of cognitive and motor deficits in both female and male rats, resulting in slower weight gain, reductions in TH expression, pronounced pathological damage to SNpc (substantia nigra pars compacta) neurons, and higher levels of Iba1 microglial activation (Zhu et al., 2022). These findings are similar to those observed in cases of atypical Parkinsonism.
Small noncoding RNAs (sncRNAs) play a range of roles in cellular function in physiological and pathological settings, governing proliferative, migratory, and apoptotic activities through the regulation of mRNA stability, transposons, transcription, translation, and reverse transcriptional processes. One postmortem study revealed that 125 microRNAs (miRNAs) were differentially expressed in prefrontal cortex tissue samples from PD patients relative to those from controls (Hoss et al., 2016). Magee et al. determined that PD patients could also be differentiated from healthy control individuals based on analyses of transfer RNA-derived small RNA (tsRNA) expression in serum, cerebrospinal fluid, and prefrontal cortex samples, thus suggesting that these tsRNAs may offer value as predictive biomarkers associated with this debilitating disease (Magee et al., 2019). Early-onset PD is closely linked to the transposable element (TE) recombination events, and TE dysregulation was found to be driven in part by pathogenic tau-induced piRNA depletion in studies of Drosophila and postmortem human tissue samples (Sun et al., 2018). Based on these results, the present study was conducted under the hypothesis that sncRNAs are similarly dysregulated and/or functionally important in the pathogenesis of manganism.
Many studies in recent years have successfully leveraged high-throughput RNA sequencing (RNA-Seq) strategies to discover the functions of particular sncRNAs. As tsRNAs and ribosomal RNA-derived small RNAs (rsRNAs) exhibit high levels of modification, including 3’-terminal 3’-phosphate and 2’,3’-cyclic phosphate modifications that interfere with the process of adapter ligation as well as m1A, m3C, m1G, and m22G methylation that disrupts reverse transcription, they are often overlooked in traditional RNA-Seq studies. To overcome this issue, Shi et al. recently designed the PANDORA-Seq approach as a means of detecting modified sncRNAs that go undetected in traditional RNA-Seq studies (Shi et al., 2021).
Here, the PANDORA-Seq approach was used to identify sncRNAs associated with manganism in rats. In total, 599 sncRNAs were found to be differentially expressed in the striatum of rats treated with MMT relative to control rats, while 1155 sncRNAs in positive control Mn-treated rats were differentially expressed relative to control animals. Subsequent functional and predictive analyses ultimately supported a potential role for sncRNA dysregulation in the pathogenesis of manganism in rats.
MATERIALS AND METHODS
Animal treatments
The ARRIVE guidelines were used to guide all animal studies. Rats were housed under controlled conditions (22 ± 2°C, 455 ± 10% relative humidity, 12 hr light/dark cycle) with free food and water access. Rat treatments were implemented as in a prior study (Zhu et al., 2022). Briefly, following a 1-week acclimation period, 18 total rats (220 ± 20 g, 8 weeks old; 9 males, 9 females) were randomly assigned to three groups (n = 3/group/sex): control, MMT, and positive control (MnCl2) groups. Animals in the MMT group were intragastrically (i.g.) administered 4 mg/kg MMT (~1 mg Mn/kg) in corn oil once per day, 6 days a week for 8 weeks. Negative control rats were administered 1 mL/kg of corn oil on the same schedule, while rats in the positive control group were administered 200 mg/kg MnCl2·4H2O (i.g.) in physiological saline (10 mL/kg). All animals were weighed daily.
Tissue preparation
Individual rats were euthanized, and samples of striatum tissue were harvested and frozen in liquid nitrogen.
RNA Isolation and Treatment
TRIzol (1 mL; Invitrogen; 15596018) was used to extract RNA from 50-100 mg of pulverized tissue samples. Following solubilization at room temperature for 5 min, 200 μL of chloroform was added to each sample and the tubes were vortexed for 15 sec, incubated at room temperature for 2-3 min, and centrifuged (10,000 xg, 10 min, 4°C). The clear upper phase was then transferred to a new tube and combined with an equal amount of isopropanol for 10 min, followed by centrifugation (10,000 xg, 10 min, 4°C). The supernatant was discarded and the precipitate was rinsed with 1 mL of 75% ethanol, after which it was centrifuged (7,500 xg, 5 min, 4°C). The supernatant was again discarded, the precipitate was allowed to air dry for 5 min, and it was then resuspended in nuclease-free water.
Individual RNA samples (200 ng) were then prepared in a 50 μL volume containing 50 mM HEPES (pH 8.0), 75 μM ferrous ammonium sulfate (pH 5.0), 2 mM sodium ascorbate, 1 mM α-ketoglutaric acid, 50 mg/L bovine serum albumin, 4 μg/mL AlkB(Epibiotek; R1822), and 2,000 U/mL of RNase inhibitor. These reactions were then incubated for 30 min at 37°C, after which RNA was isolated as above using 500 μL of TRIzol. RNA was then further incubated for 20 min at 37°C in a 50 μL volume containing 5 μL 10 × PNK buffer (New England Biolabs; B0201S), 10 mM ATP (New England Biolabs; P0756S), 10 U T4PNK (New England Biolabs; M0201L) at 37°C for 20 min. Then, the RNA was isolated with 500 μL TRIzol reagent again.
Small RNA library construction and deep sequencing
RNA samples were separated via polyacrylamide gel electrophoresis and RNA fragments 15-45 nucleotides in length were recovered for analysis and ligated with adapters from the QIAseq® miRNA Library Kit (QIAGEN; 331505). A Bioptic Qsep100 Analyzer was used for quality control, and amplified flow cell sequencing was conducted using an SE75 approach on an Illumina platform by Guangzhou Epibiotek Co., Ltd. (Guangzhou, China).
Small RNA-seq data quality control
Sequencing data were subjected to standard quality control processes to remove reads meeting the following criteria: (1) N-containing reads, (2) reads containing at least 5 bases with a quality score of < 10, (3) reads containing at least 7 bases with a quality score of < 13, (4) reads lacking an insert tag, (5) poly-A-containing reads, (6) reads lacking 3’ primers of with 5’ primer contamination, and (7) reads < 15 or > 44 nucleotides in length. The resultant clean sequencing data were retained for further analyses.
Small RNA annotation and PANDORA-seq data analyses
Adaptor sequences and low-quality reads were removed with Trimmomatic (Bolger et al., 2014), and clean reads were then aligned to miRBase 22 sequences (Kozomara et al., 2019). Those reads that remained unmapped and were 24-33 bp long were aligned to the piRNA Cluster database (Rosenkranz, 2016), piRNA sequences from NCBI, and the GtRNA database for piRNA-derived tRNAs (Chan and Lowe, 2016). Following rRNA filtering, the remaining unmapped reads were aligned to tsRBase (Zuo et al., 2021), and rsRNA alignment and annotation were performed with SPORTS 1.1 (Shi et al., 2018) using the pre-compiled annotation database.
Analyses of differentially expressed sncRNAs
The R (v 1.30.0) DEGseq package was used for pairwise comparisons of differentially expressed (DE) sncRNAs (Wang et al., 2010), with log2(fold change [FC]) > 1 and P < 0.05 as the criteria for significance. Differential rsRNA analyses were performed with SPORTS 1.1.
Target prediction, functional enrichment analysis, and hub gene network analysis
Candidate target genes of the identified DE-sncRNAs were predicted using miRanda v3.3a (Enright et al., 2003) and RNAhybrid (Krüger and Rehmsmeier, 2006), with significant target genes being estimated using selected cutoff criteria (score > 150 and energy < -20 for miRanda; energy < -25 for RNAhybird). Rat (Rattus Novegicus) 3’UTR sequences were downloaded from ensembl.org using the biomaRt R package (Durinck et al., 2009). Predicted target genes identified using these approaches were then subjected to annotation and functional enrichment analyses for both Gene Ontology (GO) biological processes and KEGG pathways using the clusterProfiler package (v 4.0.5) (Wu et al., 2021). Hub genes were identified by constructing a network of the predicted targets of DE-sncRNAs from the MMT-treated vs. control comparison in Cytoscape 3.9.0 (Shannon et al., 2003) using MCODE (Bader and Hogue, 2003) and StringApp (Doncheva et al., 2019), filtered with nervous system > 3.0.
GEO analyses
A Gene Expression Omnibus (GEO) dataset (GSE153017) comprised of rat striatum samples was used to assess how many of the sncRNAs identified in this study are expressed in rat striatum tissue. In addition, two GEO datasets (GSE71968 and GSE150646) consisting of high-throughput sequencing data from rat models of PD were analyzed to compare the targets of DE-sncRNAs from this study to known PD-related genes. The R DESeq2 package was used to conduct differential expression analyses for each GEO dataset using the following significance criteria: log2(FC) > 0.5 and P < 0.05.
RESULTS
PANDORA-seq analyses of sncRNA expression in manganese-exposed rats
Initially, rats were exposed to MMT or manganese for 8 weeks. Following this exposure period, RNA was isolated from striatum tissue samples from exposed or age-matched control rats and subjected to T4PNK and AlkB treatment. The 15- to 45- nucleotide sequences were then separated and used for sncRNA sequencing. The distribution of these sncRNA sequences revealed that rsRNAs were highly expressed in all groups, that miRNAs peaked at 22 nt in length, and that rsRNAs peaked at 16 and 81 nt in length (Fig. 1A). Identified rsRNAs were primarily derived from 18S, 28S, and 5.8S rRNA sequences (Fig. 1B), while tsRNAs were annotated into the 3’tRF, 3’tRH, 5’tRF, 5’tRH, and intertRF subtypes using tsRBase. Of these, intertRF sequences were the most abundant, whereas 5’tRF and 5’tRH sequences were the least abundant, and intermediate levels of 3’tRF expression were detected (Fig. 1C).
Identification of differential expressed sncRNAs
In total, 105 and 494 sncRNAs were upregulated and downregulated, respectively, in striatum tissue samples from MMT-treated rats relative to controls (Fig. 2A and Table S1). Of the analyzed sncRNA subtypes, tsRNAs exhibited the highest levels of variability with 68 and 362 being, respectively, up- and downregulated. When comparing Mn-treated and control rats, 426 and 729 sncRNAs were, respectively, up- and downregulated in striatum samples (Fig. 2B and Table S2), including 189 upregulated and 517 downregulated rsRNAs, as well as 76 upregulated and 208 downregulated tsRNAs. The R pheatmap package was then used to construct heatmaps corresponding to differential patterns of sncRNA expression for the MMT-treated vs. control and Mn-treated vs. control comparisons, revealing particularly pronounced differences in downregulated sncRNAs relative to upregulated sncRNAs (Fig. 2C). A Venn diagram was further constructed to highlight the overlap between DE-sncRNAs identified in the MMT-treated vs. control and Mn-treated vs. control comparisons (Fig. 2D).

As miRNAs are known to suppress the translation of target mRNAs, this mechanism may represent one key pathway through which sncRNAs function (Kumar et al., 2014; Wei et al., 2013; Zhang et al., 2015). Accordingly, potential target genes of these DE-sncRNAs identified for the MMT-treated vs. control and Mn-treated vs. control comparisons were next identified based on the overlap between the RNAHybrid and miRanda algorithms, leading to the identification of 5,872 predicted targets of DE-sncRNAs for the MMT-treated vs. control comparison (Table S3), and 9,575 for the Mn-treated vs. control comparison (Table S4). Of these targets, 5,033 genes overlapped between these two comparisons (Fig. 3A), including Drd1, Drd2, Gba, Peak1, and Mapk10. Analysis of the different types of DE-sncRNAs identified in this study also led to the identification of candidate targets of DE-tsRNA, DE-rsRNA, DE-piRNA, and DE-miRNA transcripts (Fig. 3B), with some overlap among the targets of these different sncRNAs (Fig. 3C and S1). For full list of these predicted targets, see Tables S3 and S4.

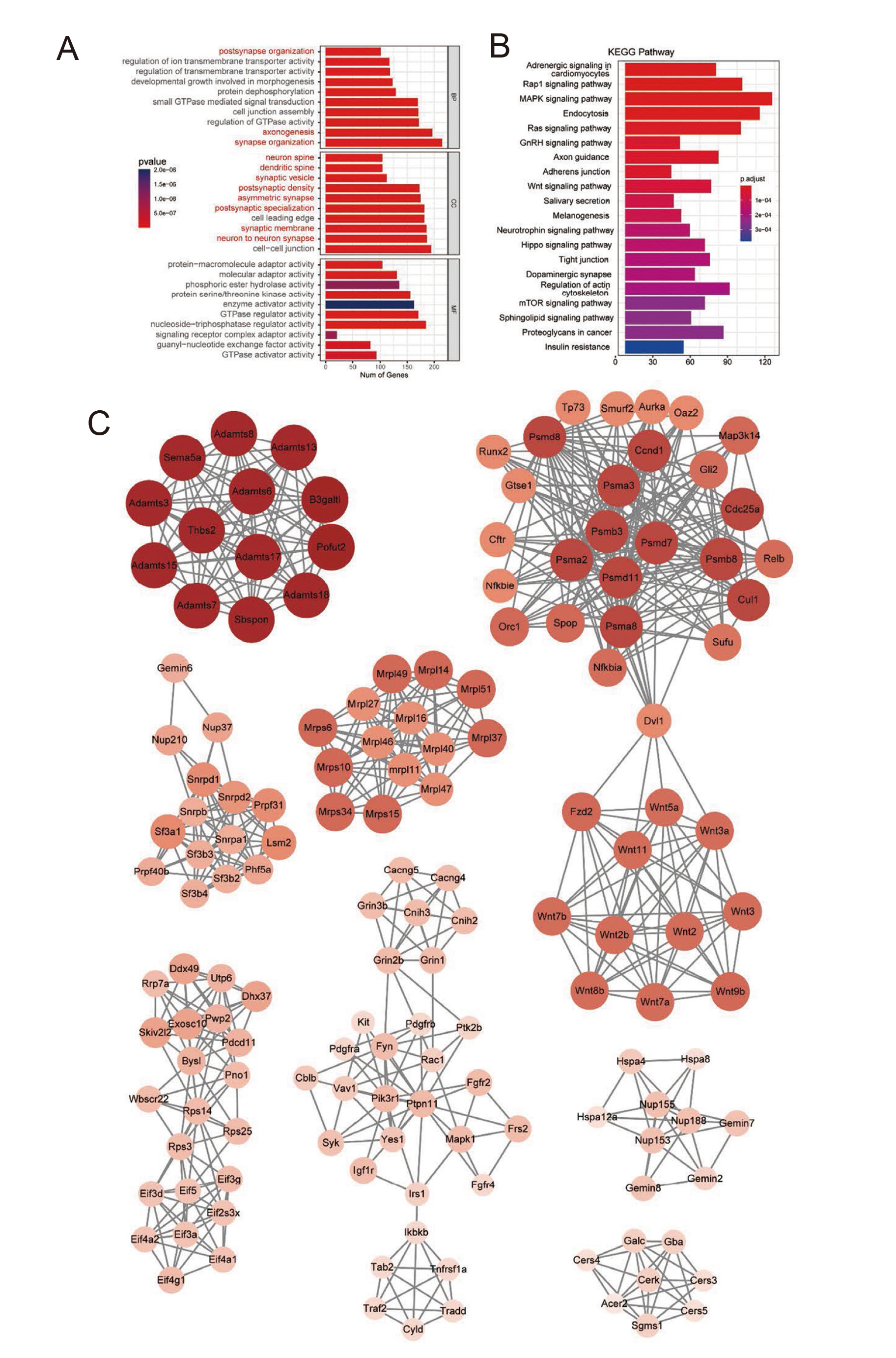
To understand the potential mechanistic functions of the identified targets of DE-sncRNAs, GO and KEGG enrichment analyses were next conducted. The targets of DE-sncRNAs for the MMT-treated vs. control comparison were enriched in several neuron-related GO terms including “postsynapse organization”, “axonogenesis”, “synapse organization”, “neuron spine”, “dendritic spine”, “synaptic vesicle”, “postsynaptic density”, and “neuron to neuron synapse” (Fig. 4A). Predicted tsRNA targets were primarily enriched in terms including “Rap1 signaling pathway”, “MAPK signaling pathway”, “Endocytosis”, and “Ras signaling pathway” (Fig. 4B). To assess which sncRNAs and target genes thereof may play the most functionally important roles in the striatum, Cytoscape was used to conduct interaction analyses for these sncRNAs and their predicted targets. The top 20 sncRNAs sorted by degree value in this analysis included rno_tsr007375, pir-64524@piRNAcluster, rno_tsr007374, rno_tsr007372, rno_tsr006430, rno_tsr002254, rno_tsr005113, rno_tsr001016, rno_tsr007373, rno_tsr005444, rno_tsr000168, rno_tsr003352, rno-miR-615, rno_tsr003812, rno_tsr009096, rno_tsr009697, rno_tsr006240, rno_tsr005242, rsRNA05025, rno_tsr009696 (Fig. S1 and Table S5). The STRING database was further used to identify predicted hub target genes potentially related to neurotoxicity, including Adamts, Wnts, Mrps/Mrpl, Eif-Rps, Mapk, Hsp, Gba, and Vamp2 (Fig. 4C).
To finally confirm the relevance of the predicted sncRNA target genes in striatum samples from rats, these datasets were compared with the GSE153017 dataset comprised of transcriptomic data from rat striatum tissue samples. This analysis revealed that ~80% of the predicted target genes were represented in that GEO dataset, thus confirming that the majority of these genes are truly expressed in the striatum (Fig. 5A). While relatively few predicted target genes for the MMT-treated vs. control comparison overlapped with GEO datasets derived from two different rat models of PD, certain important PD-related genes did overlap between these datasets including Park7, Mapk10, Casp3, and Casp9 (Fig. 5B). The GSE71968 dataset was comprised of RNA-Seq data from frontal cortex tissue samples isolated from untreated rats and rats overexpressing human SNCA, while the GSE150646 dataset was comprised of data from the brains of wild-type and DJ-1 knockout rats.
DISCUSSION
In this study, two miRNAs were identified as being related to manganism in rats, which have also been reported in prior research studies. For example, the upregulation of miR-200a has been reported in PC12 and SH-SY5Y cells in which oxidative stress was induced by MPP+ exposure (Salimian et al., 2018; Talepoor Ardakani et al., 2019). Inhibiting miR-200a can reportedly suppress apoptotic cell death and increase the size of the dopaminergic neuron population in the 6-OHDA-treated rat model of PD (Wu et al., 2018). Moreover, the inflammation-related miRNA miR-146a has been identified as among the most significantly upregulated miRNAs in a rotenone-induced rat model of PD (Jauhari et al., 2020), with miR-146a in turn being responsible for the downregulation of Parkin protein levels. Inhibiting miR-146a was sufficient to reduce NF-κβ phosphorylation and to enhance Parkin expression in SH-SY5Y cells treated with rotenone (Jauhari et al., 2020). While other studies have explored neurological disease-related changes in piRNAs (Schulze et al., 2018; Sun et al., 2018) or tsRNAs (Magee et al., 2019), comparing these prior findings to the present results was difficult due to species differences and differences in the utilized database types. While some rsRNAs have been reportedly linked to diabetes (Wei et al., 2013), no studies have, to our knowledge, previously documented the association between rsRNAs and PD or manganism.
Here, the miRanda and RNAHybrid tools were used to predict DE-sncRNA targets based on miRNA-mRNA interactions. While these algorithms were not effective when assessing tsRNAs or rsRNAs longer than 30 nt in length, some potentially interesting targets were nonetheless identified. For example, Slc39a10 and Slc39a14 are key Mn transporters, and modulation of their expression may be related to preventing Mn-induced neurotoxicity (Aydemir et al., 2017; Santini et al., 2009). Drd1 and Drd2 were targets of DE-sncRNAs identified for both the MTT-treated vs. control and Mn-treated vs. control comparisons. Adenylate cyclase binding (GO:0008179) was enriched in GO analyses for these predicted target genes, and “dopaminergic synapse” was among the top 20 most enriched KEGG pathways suggesting that manganism is related to dopaminergic dysfunction. Drd1 and Drd2 can stimulate or inhibit adenylate cyclase activity (Vallone et al., 2000). As such, exposure to Mn may result in dopaminergic dysfunction owing to the ability of specific sncRNAs to inhibit Drd1 and Drd2 translation. Slc1a3 is the primary enzyme responsible for synaptic glutamate clearance and was also identified at the intersection of predicted target genes for both the MMT-treated vs. control and Mn-treated vs. control comparisons. Glutamate receptor binding (GO:0035254) and Ionotropic glutamate receptor binding (GO:0035255) were enriched in GO analyses, and in cultured rat astrocytes lower levels of Slc1a3 expression have been reported following Mn exposure, with a concomitant drop in glutamate uptake (Erikson and Aschner, 2002). Many neuron- and GTPase-related GO terms were also enriched in these analyses, suggesting that sncRNAs may play a role in manganism through the modulation of neuronal GTPase activity, in line with prior work highlighting GTPases as mediators of PD pathogenesis and potential targets for pharmacological intervention (Hong and Sklar, 2014; Obergasteiger et al., 2018).
Rap1 and Ras are small GTPases, and the Rap1 and Ras signaling pathways were highly enriched in KEGG analyses of the predicted target genes of the identified DE-sncRNAs. The MAPK, NF-κB, and PI3K-Akt pathways are all closely related to reactive oxygen species (ROS) production, which is notable given that many studies have detailed the link between Mn exposure and oxidative stress (Tinkov et al., 2021). Melanogenesis was also represented in KEGG enrichment pathways. Neuromelanin normally serves as a protective metal chelator within neurons, thus mitigating oxidative stress-related damage. When active metal and toxicant concentrations exceed a particular threshold, however, neuromelanin loses its antioxidant potential and instead functions in a pro-oxidative manner through a feedback loop that ultimately exacerbates cellular oxidative stress (Moreno-García et al., 2021). GABA can influence the switching of gonadotropin-releasing hormone (GnRH) neurons from a depolarized to a hyperpolarized state in female mice (Han et al., 2002), and GnRH is an important regulator of neuromodulation in the context of vertebrate reproduction. Exposure to Mn results in abnormal GnRH secretion (Wu et al., 2020). Neurotrophins, HIPPO, and sphingolipids have all been implicated in neurodegeneration (Alessenko and Albi, 2020; Lin et al., 2019; Mitre et al., 2017; Mueller et al., 2018), and were all enriched in KEGG analyses of DE-sncRNA target genes.
Hub gene analyses of predicted targets of the DE-sncRNAs identified for the MMT-treated vs. control comparison led to the identification of Adamts (Elsadany et al., 2021), Mrps/Mrpl (Babenko et al., 2018), Eif-Rps (Deng et al., 2015), MAPK (Bohush et al., 2018), Gba (Sidransky and Lopez, 2012), Vamps2 (Vallortigara et al., 2016), Hsp, and Wnts, which were included in the Harmonizome PD gene set (Rouillard et al., 2016), with all of these genes being related to neurodegenerative disease. Some of these target genes also overlapped with published GEO datasets of genes from rat models of PD, including key neurotoxicity-related genes including Park7 (Xu et al., 2010), Mapk10 (Crittenden and Filipov, 2011), Casp3, and Casp9 (Tarale et al., 2016).
While few bioinformatics sources and databases are available for these analyses, PANDORA-Seq was nonetheless a powerful approach to analyzing sncRNAs, while SPORTS 1.1 was able to facilitate the annotation of rsRNAs without access to an rsRNA database.
In conclusion, the PANDORA-Seq tool was herein used to identify sncRNAs that are dysregulated in unrepaired striatal tissue from MMT-treated rats. Through further functional analyses of the predicted targets of these DE-sncRNAs, these findings highlight a potentially critical role for these sncRNAs as coordinators of the pathophysiology of manganism in rats through the regulation of a variety of genes, including important neurotoxicity-related targets.
ACKNOWLEDGMENTS
This work was supported by the Technology Innovation Guidance Project of Guangxi, China [Grant No. 2018AC01003], Guangxi Science and Technology Base and Talent Special Projects [Grant No. 2021AC18006], Guangxi Key Research and Development Program [Grant No. 2021AB09011].
Conflict of interest
The authors declare that there is no conflict of interest.
REFERENCES
- Alessenko, A.V. and Albi, E. (2020): Exploring sphingolipid implications in neurodegeneration. Front. Neurol., 11, 437.
- Aschner, M. and Aschner, J.L. (1991): Manganese neurotoxicity: cellular effects and blood-brain barrier transport. Neurosci. Biobehav. Rev., 15, 333-340.
- Aydemir, T.B., Kim, M.H., Kim, J., Colon-Perez, L.M., Banan, G., Mareci, T.H., Febo, M. and Cousins, R.J. (2017): Metal transporter Zip14 (Slc39a14) deletion in mice increases manganese deposition and produces neurotoxic signatures and diminished motor activity. J. Neurosci., 37, 5996-6006.
- Babenko, V.N., Smagin, D.A., Galyamina, A.G., Kovalenko, I.L. and Kudryavtseva, N.N. (2018): Altered Slc25 family gene expression as markers of mitochondrial dysfunction in brain regions under experimental mixed anxiety/depression-like disorder. BMC Neurosci., 19, 79.
- Bader, G.D. and Hogue, C.W. (2003): An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics, 4, 2.
- Bakthavatsalam, S., Das Sharma, S., Sonawane, M., Thirumalai, V. and Datta, A. (2014): A zebrafish model of manganism reveals reversible and treatable symptoms that are independent of neurotoxicity. Dis. Model. Mech., 7, 1239-1251.
- Bohush, A., Niewiadomska, G. and Filipek, A. (2018): Role of mitogen activated protein kinase signaling in parkinson’s disease. Int. J. Mol. Sci., 19, 2973.
- Bolger, A.M., Lohse, M. and Usadel, B. (2014): Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114-2120.
- Calne, D.B., Chu, N.S., Huang, C.C., Lu, C.S. and Olanow, W. (1994): Manganism and idiopathic parkinsonism: similarities and differences. Neurology, 44, 1583-1586.
- Chan, P.P. and Lowe, T.M. (2016): GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res., 44 (D1), D184-D189.
- Couper, J. (1837): On the effects of black oxide of manganese when inhaled into the lungs. Br. Ann. Med. Pharmacol., 1, 42-43.
- Crittenden, P.L. and Filipov, N.M. (2011): Manganese modulation of MAPK pathways: effects on upstream mitogen activated protein kinase kinases and mitogen activated kinase phosphatase-1 in microglial cells. J. Appl. Toxicol., 31, 1-10.
- Deng, H., Wu, Y. and Jankovic, J. (2015): The EIF4G1 gene and Parkinson’s disease. Acta Neurol. Scand., 132, 73-78.
- Doncheva, N.T., Morris, J.H., Gorodkin, J. and Jensen, L.J. (2019): Cytoscape StringApp: network analysis and visualization of proteomics data. J. Proteome Res., 18, 623-632.
- Dorman, D.C., McManus, B.E., Marshall, M.W., James, R.A. and Struve, M.F. (2004): Old age and gender influence the pharmacokinetics of inhaled manganese sulfate and manganese phosphate in rats. Toxicol. Appl. Pharmacol., 197, 113-124.
- Durinck, S., Spellman, P.T., Birney, E. and Huber, W. (2009): Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc., 4, 1184-1191.
- Elsadany, M., Elghaish, R.A., Khalil, A.S., Ahmed, A.S., Mansour, R.H., Badr, E. and Elserafy, M. (2021): Transcriptional analysis of nuclear-encoded mitochondrial genes in eight neurodegenerative disorders: the analysis of seven diseases in reference to Friedreich’s ataxia. Front. Genet., 12, 749792.
- Enright, A.J., John, B., Gaul, U., Tuschl, T., Sander, C. and Marks, D.S. (2003): MicroRNA targets in Drosophila. Genome Biol., 5, R1.
- Erikson, K. and Aschner, M. (2002): Manganese causes differential regulation of glutamate transporter (GLAST) taurine transporter and metallothionein in cultured rat astrocytes. Neurotoxicology, 23, 595-602.
- Han, S.K., Abraham, I.M. and Herbison, A.E. (2002): Effect of GABA on GnRH neurons switches from depolarization to hyperpolarization at puberty in the female mouse. Endocrinology, 143, 1459-1466.
- Hong, L. and Sklar, L.A. (2014): Targeting GTPases in Parkinson’s disease: comparison to the historic path of kinase drug discovery and perspectives. Front. Mol. Neurosci., 7, 52.
- Hoss, A.G., Labadorf, A., Beach, T.G., Latourelle, J.C. and Myers, R.H. (2016): microRNA profiles in Parkinson’s disease prefrontal cortex. Front. Aging Neurosci., 8, 36.
- Jauhari, A., Singh, T., Mishra, S., Shankar, J. and Yadav, S. (2020): Coordinated action of miR-146a and parkin gene regulate rotenone-induced neurodegeneration. Toxicol. Sci., 176, 433-445.
- Kozomara, A., Birgaoanu, M. and Griffiths-Jones, S. (2019): miRBase: from microRNA sequences to function. Nucleic Acids Res., 47 (D1), D155-D162.
- Krüger, J. and Rehmsmeier, M. (2006): RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res., 34, W451-4.
- Kulshreshtha, D., Ganguly, J. and Jog, M. (2021): Manganese and Movement Disorders: A Review. J. Mov. Disord., 14, 93-102.
- Kumar, P., Anaya, J., Mudunuri, S.B. and Dutta, A. (2014): Meta-analysis of tRNA derived RNA fragments reveals that they are evolutionarily conserved and associate with AGO proteins to recognize specific RNA targets. BMC Biol., 12, 78.
- Kwakye, G.F., Paoliello, M.M., Mukhopadhyay, S., Bowman, A.B. and Aschner, M. (2015): Manganese-induced parkinsonism and Parkinson’s disease: shared and distinguishable features. Int. J. Environ. Res. Public Health, 12, 7519-7540.
- Lin, G., Wang, L., Marcogliese, P.C. and Bellen, H.J. (2019): Sphingolipids in the pathogenesis of Parkinson’s disease and parkinsonism. Trends Endocrinol. Metab., 30, 106-117.
- Lu, C.-S., Huang, C.-C., Chu, N.-S. and Calne, D.B. (1994): Levodopa failure in chronic manganism. Neurology, 44, 1600-1602.
- Magee, R., Londin, E. and Rigoutsos, I. (2019): TRNA-derived fragments as sex-dependent circulating candidate biomarkers for Parkinson’s disease. Parkinsonism Relat. Disord., 65, 203-209.
- Mitre, M., Mariga, A. and Chao, M.V. (2017): Neurotrophin signalling: novel insights into mechanisms and pathophysiology. Clin. Sci. (Lond.), 131, 13-23.
- Moreno-García, A., Kun, A., Calero, M. and Calero, O. (2021): The neuromelanin paradox and its dual role in oxidative stress and neurodegeneration. Antioxidants, 10, 1-19.
- Mueller, K.A., Glajch, K.E., Huizenga, M.N., Wilson, R.A., Granucci, E.J., Dios, A.M., Tousley, A.R., Iuliano, M., Weisman, E., LaQuaglia, M.J., DiFiglia, M., Kegel-Gleason, K., Vakili, K. and Sadri-Vakili, G. (2018): Hippo signaling pathway dysregulation in human Huntington’s disease brain and neuronal stem cells. Sci. Rep., 8, 11355.
- Obergasteiger, J., Frapporti, G., Pramstaller, P.P., Hicks, A.A. and Volta, M. (2018): A new hypothesis for Parkinson’s disease pathogenesis: GTPase-p38 MAPK signaling and autophagy as convergence points of etiology and genomics. Mol. Neurodegener., 13, 40.
- Perl, D.P. and Olanow, C.W. (2007): The neuropathology of manganese-induced Parkinsonism. J. Neuropathol. Exp. Neurol., 66, 675-682.
- Racette, B.A. (2014): Manganism in the 21st century: the Hanninen lecture. Neurotoxicology, 45, 201-207.
- Röllin, H.B. and Nogueira, C.M. (2011): Manganese: environmental pollution and health effects, in: encyclopedia of environmental health. Elsevier, pp. 617-629.
- Rosenkranz, D. (2016): piRNA cluster database: a web resource for piRNA producing loci. Nucleic Acids Res., 44 (D1), D223-D230.
- Rouillard, A.D., Gundersen, G.W., Fernandez, N.F., Wang, Z., Monteiro, C.D., McDermott, M.G. and Ma’ayan, A. (2016): The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database (Oxford), 2016, baw100.
- Salimian, N., Peymani, M., Ghaedi, K. and Nasr Esfahani, M.H. (2018): Modulation in miR-200a/SIRT1axis is associated with apoptosis in MPP+-induced SH-SY5Y cells. Gene, 674, 25-30.
- Santini, E., Heiman, M., Greengard, P., Valjent, E. and Fisone, G. (2009): Inhibition of mTOR signaling in Parkinson’s disease prevents L-DOPA-induced dyskinesia. Sci. Signal., 2, ra36.
- Scholten, J.M. (1953): On manganese encephalopathy; description of a case. Folia Psychiatr. Neurol. Neurochir. Neerl., 56, 878-884.
- Schulze, M., Sommer, A., Plötz, S., Farrell, M., Winner, B., Grosch, J., Winkler, J. and Riemenschneider, M.J. (2018): Sporadic Parkinson’s disease derived neuronal cells show disease-specific mRNA and small RNA signatures with abundant deregulation of piRNAs. Acta Neuropathol. Commun., 6, 58.
- Shannon, P., Markiel, A., Ozier, O., Baliga, N.S., Wang, J.T., Ramage, D., Amin, N., Schwikowski, B. and Ideker, T. (2003): Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res., 13, 2498-2504.
- Shi, J., Ko, E.A., Sanders, K.M., Chen, Q. and Zhou, T. (2018): SPORTS1.0: A tool for annotating and profiling non-coding RNAs optimized for rRNA- and tRNA-derived small RNAs. Genomics Proteomics Bioinformatics, 16, 144-151.
- Shi, J., Zhang, Y., Tan, D., Zhang, X., Yan, M., Zhang, Y., Franklin, R., Shahbazi, M., Mackinlay, K., Liu, S., Kuhle, B., James, E.R., Zhang, L., Qu, Y., Zhai, Q., Zhao, W., Zhao, L., Zhou, C., Gu, W., Murn, J., Guo, J., Carrell, D.T., Wang, Y., Chen, X., Cairns, B.R., Yang, X.L., Schimmel, P., Zernicka-Goetz, M., Cheloufi, S., Zhang, Y., Zhou, T. and Chen, Q. (2021): PANDORA-seq expands the repertoire of regulatory small RNAs by overcoming RNA modifications. Nat. Cell Biol., 23, 424-436.
- Sidransky, E. and Lopez, G. (2012): The link between the GBA gene and parkinsonism. Lancet Neurol., 11, 986-998.
- Sun, W., Samimi, H., Gamez, M., Zare, H. and Frost, B. (2018): Pathogenic tau-induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat. Neurosci., 21, 1038-1048.
- Talepoor Ardakani, M., Rostamian Delavar, M., Baghi, M., Nasr-Esfahani, M.H., Kiani-Esfahani, A. and Ghaedi, K. (2019): Upregulation of miR-200a and miR-204 in MPP+ -treated differentiated PC12 cells as a model of Parkinson’s disease. Mol. Genet. Genomic Med., 7, e548.
- Tarale, P., Chakrabarti, T., Sivanesan, S., Naoghare, P., Bafana, A. and Krishnamurthi, K. (2016): Potential role of epigenetic mechanism in manganese induced neurotoxicity. BioMed Res. Int., 2016, 2548792.
- Tinkov, A.A., Paoliello, M.M., Mazilina, A.N., Skalny, A.V., Martins, A.C., Voskresenskaya, O.N., Aaseth, J., Santamaria, A., Notova, S.V., Tsatsakis, A., Lee, E., Bowman, A.B. and Aschner, M. (2021): Molecular targets of manganese-induced neurotoxicity: A five-year update. Int. J. Mol. Sci., 22, 4646.
- Vallone, D., Picetti, R. and Borrelli, E. (2000): Structure and function of dopamine receptors. Neurosci. Biobehav. Rev., 24, 125-132.
- Vallortigara, J., Whitfield, D., Quelch, W., Alghamdi, A., Howlett, D., Hortobágyi, T., Johnson, M., Attems, J., O’Brien, J.T., Thomas, A., Ballard, C.G., Aarsland, D. and Francis, P.T. (2016): Decreased levels of VAMP2 and monomeric alpha-synuclein correlate with duration of dementia. J. Alzheimers Dis., 50, 101-110.
- Wang, L., Feng, Z., Wang, X., Wang, X. and Zhang, X. (2010): DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics, 26, 136-138.
- Wei, H., Zhou, B., Zhang, F., Tu, Y., Hu, Y., Zhang, B. and Zhai, Q. (2013): Profiling and identification of small rDNA-derived RNAs and their potential biological functions. PLoS One, 8, e56842.
- Wolters, E.C., Huang, C.-C., Clark, C., Peppard, R.F., Okada, J., Chu, N.-S., Adam, M.J., Ruth, T.J., Li, D. and Calne, D.B. (1989): Positron emission tomography in manganese intoxication. Ann. Neurol., 26, 647-651.
- Wu, D.M., Wang, S., Wen, X., Han, X.R., Wang, Y.J., Shen, M., Fan, S.H., Zhuang, J., Zhang, Z.F., Shan, Q., Li, M.Q., Hu, B., Sun, C.H., Lu, J. and Zheng, Y.L. (2018): Inhibition of microRNA-200a upregulates the expression of striatal dopamine receptor d2 to repress apoptosis of striatum via the cAMP/PKA signaling pathway in rats with Parkinson’s disease. Cell. Physiol. Biochem., 51, 1600-1615.
- Wu, F., Yang, H., Liu, Y., Yang, X., Xu, B., Liu, W., Xu, Z. and Deng, Y. (2020): Manganese exposure caused reproductive toxicity of male mice involving activation of GnRH secretion in the hypothalamus by prostaglandin E2 receptors EP1 and EP2. Ecotoxicol. Environ. Saf., 201, 110712.
- Wu, T., Hu, E., Xu, S., Chen, M., Guo, P., Dai, Z., Feng, T., Zhou, L., Tang, W., Zhan, L., Fu, X., Liu, S., Bo, X. and Yu, G. (2021): clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation(China), 2, 100141.
- Xu, X., Martin, F. and Friedman, J.S. (2010): The familial Parkinson’s disease gene DJ-1 (PARK7) is expressed in red cells and plays a role in protection against oxidative damage. Blood Cells Mol. Dis., 45, 227-232.
- Zhang, P., Kang, J.Y., Gou, L.T., Wang, J., Xue, Y., Skogerboe, G., Dai, P., Huang, D.W., Chen, R., Fu, X.D., Liu, M.F. and He, S. (2015): MIWI and piRNA-mediated cleavage of messenger RNAs in mouse testes. Cell Res., 25, 193-207.
- Zhu, Q.F., Lu, L.L., Fang, Y.Y., Wu, J., Huang, Z.Y., Zheng, X.W., Song, H.-X., Aschner, M., Song, C. and Jiang, Y.M. (2022): Methylcyclopentadienyl manganese tricarbonyl alter behavior and cause ultrastructural changes in the substantia nigra of rats: comparison with inorganic manganese chloride. Neurochem. Res., 47, 2198-2210.
- Zuo, Y., Zhu, L., Guo, Z., Liu, W., Zhang, J., Zeng, Z., Wu, Q., Cheng, J., Fu, X., Jin, Y., Zhao, Y. and Peng, Y. (2021): tsRBase: a comprehensive database for expression and function of tsRNAs in multiple species. Nucleic Acids Res., 49 (D1), D1038-D1045.

