Abstract
Methylmercury (MeHg), an environmental pollutant, disrupts and impairs cellular function. MeHg binds to various cellular proteins, causing dysfunction and misfolding, which are considered underlying causes of MeHg toxicity. The p62 protein, also termed SQSTM1, is a ubiquitin-binding protein that targets ubiquitinated substrates to undergo autophagy and plays a key role in ameliorating MeHg toxicity. p62 also delivers ubiquitinated substrates to proteasomes. However, the role of these degradation systems in mitigating MeHg toxicity remains unknown. Herein, we explored the impact of the proteasome inhibitor MG132 on MeHg toxicity and examined the toxicity of co-treatment with MG132 and MeHg in p62KO mouse embryonic fibroblasts (MEFs) by analyzing cell viability, immunoblotting, mRNA levels, immunofluorescence, and the mercury content. The proteasome inhibitor MG132 enhanced MeHg-induced cytotoxicity while reducing intracellular mercury levels in MEFs. Co-treatment with MG132 and MeHg markedly increased levels of p62 and ubiquitinated proteins. Furthermore, co-treatment with MG132 and MeHg reduced p62KO MEF viability compared to that of wild-type MEFs. Our findings suggest that the proteasome participates in mitigating MeHg cytotoxicity, while p62 may play an important role in transporting MeHg-induced ubiquitinated proteins to the proteasome, as well as in autophagy. Collectively, these results imply that p62, and proteasome, and autophagy are vital for cytoprotection against MeHg toxicity.
INTRODUCTION
Methylmercury (MeHg), an environmental pollutant, damages the nervous system and organs of the body (Clarkson and Magos, 2006). MeHg disrupts various physiological functions and has been associated with developmental disorders of the central nervous system in fetuses and children, as well as neurodegenerative diseases, abnormal glucose metabolism, and heart disease in adults (Yokoo et al., 2003; Auger et al., 2005; Karagas et al., 2012). MeHg reportedly binds to glutathione and proteins in cells, indicating its potential to induce oxidative stress and protein dysfunction (Unoki et al., 2018). In response, cells activate various cellular functions, such as mitogen-activated protein kinase, endoplasmic reticulum stress, and autophagy (Ke et al., 2019).
Previously, we have focused on the possible role of proteolytic systems in mitigating MeHg toxicity and found that MeHg can activate autophagy, which is vital for protection against MeHg toxicity (Takanezawa et al., 2016, 2019b, 2021). Furthermore, we have documented that p62 (also known as SQSTM1), an autophagy-related protein, mitigates MeHg toxicity by inhibiting the MeHg-induced increase in ubiquitinated protein levels (Takanezawa et al., 2017). p62 binds to ubiquitinated proteins and transports them to autophagosomes for degradation (Seibenhener et al., 2004; Liu et al., 2016). Moreover, p62 participates in various signaling pathways, including the Keap1–Nrf2 pathway (Komatsu et al., 2010), which provides protection against MeHg toxicity (Kumagai et al., 2013). These findings indicate that the autophagy–p62 axis could play an important role in mitigating MeHg toxicity.
p62 participates in the transport of ubiquitinated proteins to proteasomes and in autophagy (Liu et al., 2016). The ubiquitin–proteasome system (UPS), another proteolytic system, is a colossal proteolytic apparatus that removes unwanted proteins from the cytoplasm and nucleus and mediates general protein homeostasis and stress response (Amm et al., 2014). The protective role of the UPS against MeHg toxicity has been corroborated in yeast cells. Overexpression of a ubiquitin-conjugating enzyme Cdc34 in yeast and human cells induces resistance to MeHg toxicity (Hwang et al., 2002). Overexpression of the UPS-related ubiquitin-binding protein Rad23 in yeast cells also enhances resistance to MeHg toxicity (Hwang et al., 2005). Although these findings suggest that the UPS may participate in allaying MeHg toxicity, the role of proteasomes in mitigation MeHg toxicity in mammalian cells remains poorly understood.
Therefore, we hypothesized that the proteasome and autophagy contribute to MeHg toxicity mitigation, with the involvement of p62. To examine this hypothesis, we investigated the impact of proteasome inhibitors on MeHg toxicity and evaluated the toxicity of co-treatment with a proteasome inhibitor and MeHg in p62KO mouse embryonic fibroblasts (MEFs). Herein, we showed that proteasome as well as autophagy inhibitors could enhance MeHg cytotoxicity, and that p62 may play an important role in both processes.
MATERIALS AND METHODS
Chemicals
MeHg chloride (Tokyo Kasei, Tokyo, Japan) was dissolved in dimethyl sulfoxide (DMSO) and stored as a 25 mM stock solution. MG132 and chloroquine diphosphate (CQ) were procured from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
Immortalized p62-/- and p62+/+ MEFs (kind gifts from M. Komatsu and T. Yanagawa) were cultured as described in a previous study (Takanezawa et al., 2017). Dulbecco’s modified Eagle’s medium (D6546, Sigma-Aldrich) supplemented with 10% fetal calf serum, 0.1 mM non-essential amino acids (11140-050, Thermo Fisher Scientific, Rockford, IL, USA), 1 mM sodium pyruvate (11360-070, Thermo Fisher Scientific), 100 units/mL penicillin, 100 µg/mL streptomycin, and 292 µg/mL L-glutamine. Cell lines were maintained at 37°C in a 5% CO2 humidified atmosphere.
Immunofluorescence
Immunofluorescence analysis was performed as described in a previous study (Takanezawa et al., 2021). Cells were fixed with 4% paraformaldehyde (Nacalai Tesque, Kyoto, Japan) and permeabilized using 0.2% Triton X-100 in phosphate-buffered saline (PBS) for 10 min. Next, the cells were blocked using 1% bovine serum albumin for 1 hr and incubated with an anti-p62 antibody (1:200). After washing with PBS, samples were treated with Alexa Fluor-conjugated secondary antibody (1:1000; Thermo Fisher Scientific) for 1 hr. Cells were mounted using ProLong Glass Antifade Mountant with NucBlue (Thermo Fisher Scientific). Immunofluorescence was visualized using the FV3000 confocal laser-scanning microscope (Olympus, Tokyo, Japan). Images were processed using the FV3000 system software (FV31S-SW).
Mercury analysis
Cells cultured in a 6-well plate were lysed using 120 µL RIPA buffer, and the intercellular mercury content in 20 µL of each cell lysate was determined using the Mercury Analyzer MA-2 (Nippon Instruments Co., Tokyo, Japan), as described in a previous study (Takanezawa et al., 2019a). The total protein content was measured using the Bio-Rad DC Protein Assay kit (Bio-Rad, Hercules, CA, USA).
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from cells using the RNA extraction kit (NucleoSpin RNA kit; Macherey-Nagel, Bethlehem, PA, USA), according to the manufacturer’s protocol, and then subjected to reverse transcription using the PrimeScript RT Master Mix (Takara, Shiga, Japan). Each real-time PCR reaction consisted of 1 µL of cDNA template, 5 µL of PowerUp SYBR Green Master Mix (Thermo Fisher Scientific), and 4 pmol of forward and reverse primers on the CFX-96 thermal cycler system (Bio-Rad) for 40 cycles (95°C for 2 min, 55°C for 10 sec, and 72°C for 10 sec) after an initial 2 min of incubation at 95°C. The mRNA level of the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (Gapdh) was used for normalization. The following primers were used for PCR: sequestosome 1/p62 (p62) forward, 5′-GTGGGACAGCCAGAGGAACA-3′ and reverse, 5′-GCCCTTCCGATTCTGGCAT-3′; Gapdh forward, 5′-AAATGGTGAAGGTCGGTGTG-3′ and reverse, 5′-TGAAGGGGTCGTTGATGG-3′. Amplicon size (p62; 136 bp and Gapdh; 108 bp) and reaction specificity were confirmed by melting curve and DNA agarose gel electrophoresis. The results were evaluated using the Bio-Rad CFX Manager software (version 3.1; Bio-Rad). The quantification cycle value was recorded, and the relative expression of the target gene was determined using the 2-∆∆Ct method.
Immunoblot analysis
Cells were lysed using RIPA buffer (20 mM Tris [pH 7.4], 0.1% sodium dodecyl sulfate (SDS), 1% sodium deoxycholate, and 1% Nonidet P-40) containing a protease/phosphatase inhibitor cocktail (Cell Signaling Technology, Danvers, MA, USA, 5872S) and sonicated for 10 sec on ice. Cell lysates were resolved by SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. Membranes were incubated with the following specific primary antibodies: anti-p62 (1:1000; PM045, Medical and Biological Laboratory, Nagoya, Japan), anti-ubiquitin (1:1000; #3936, Cell Signaling Technology), anti-GAPDH (1:2000; sc-25778, Santa Cruz Biotechnology), and an HRP-conjugated anti-rabbit secondary antibody (1:8000; NA934V, GE Healthcare, Buckinghamshire, United Kingdom). Immunoblots were visualized on the Amersham Imager 680 (GE Healthcare) using enhanced chemiluminescence detection reagents. Quantification of blots was performed using ImageQuant TL (GE Healthcare).
Cell viability assay
Cell viability was measured using the CCK-8 assay, as described in previous studies (Takanezawa et al., 2019a, 2021). Briefly, cells (1 × 104 cells/well) were seeded in 96-well plates, and pretreated with 20 µM CQ or 0.625 µM MG132 for 1 hr, and treated with 1 µM MeHg for 24 hr. After treatment, tetrazolium salt WST-8 (10 µL; Dojindo, Kumamoto, Japan) was added to each well, followed by incubation at 37°C for 1 hr. The number of viable cells was quantified by measuring absorbance at 450 nm using an iMark microplate reader (Bio-Rad, Hercules, CA, USA). Each CCK-8 assay was performed in triplicate. Cell viability was calculated using the following equation:
Cell viability (% of control) = (Absorbance of MeHg treatment–absorbance of blank)/(absorbance of treatment–absorbance of blank) × 100.
Statistical analysis
Quantitative data are expressed as means ± standard deviation. Data were analyzed using Student’s t-test employing at least three independent data points. A p-value of <0.05 was deemed statistically significant (*p < 0.05 or **p < 0.01).
RESULTS AND DISCUSSION
Although MeHg exposure can damage various cell types (Wang et al., 2009; Takanezawa et al., 2016; Hiraoka et al., 2017), evidence regarding the effect of proteasome inhibition on MeHg-induced cell death remains scarce. Herein, we first examined whether proteasome inhibition with MG132 affects p62 expression in MEF cells and determined that concentration of 0.625 µM MG132 was a good choice for this study (Supplementary Fig. 1). Based on morphological analysis by phase contrast microscopy, we observed that co-treatment with MeHg and MG132, a proteasome inhibitor, increased MEF shrinkage and aggregation and cell surface detachment as compared with MeHg or MG132 treatment alone (Fig. 1A). These findings suggest that proteasomes may protect cells from MeHg toxicity. MeHg toxicity is considerably associated with increased intracellular Hg concentrations (Usuki et al., 2017). Therefore, we examined the effect of MG132 on intracellular Hg concentration. Unexpectedly, time-course Hg measurements revealed that MG132 treatment significantly reduced intracellular Hg concentration (Fig. 1B). A decrease in intracellular mercury concentrations was observed at an early time point (6 hr) after treatment with MG132, when cell viability was not significantly reduced (Supplementary Fig. 2). Thus, the potentiating effect of MG132 on MeHg-induced cell death was independent of the increased intracellular Hg concentration.

Next, we examined the effect of MG132 on p62 expression and distribution, given that MG132 increases both p62 mRNA and protein levels in hypothalamic organotypic cultures (Tominaga et al., 2017), and MeHg can induce p62 expression in MEFs (Takanezawa et al., 2017, 2021). Notably, MG132 induced the highest p62 transcript level at 8 hr, maintaining a relatively high level for 24 hr. Combined MeHg and MG132 treatment induced high p62 expression in MEFs compared to that induced by MeHg or MG132 alone (Fig. 2A), suggesting that MG132 promotes MeHg-induced p62 expression. p62 has been shown to be upregulated by a key antioxidant pathway, the Keap1–Nrf2 system (Liu et al., 2016), and MG132 and MeHg can activate Nrf2 (Unoki et al., 2018). Therefore, the increased expression of p62 by combined MeHg and MG132 treatment was suggested to be due to the activation of Nrf2 rather than by the treatment of each alone. Consistent with p62 mRNA induction, co-treatment with MG132 and MeHg increased p62 protein levels (Fig. 2B, Upper). In line with p62 protein induction, ubiquitinated proteins accumulated in MeHg- and MG132-treated cells. Ubiquitinated proteins accumulated faster (after 2 hr after treatment) in MG132 and MeHg co-treated cells than in MG132-treated cells. Accordingly, the UPS is necessary, at least partly, for removing MeHg-induced ubiquitinated proteins. However, we noted minimal differences in ubiquitinated protein accumulation between MeHg-treated and MeHg and MG132-co-treated cells after 24 hr (Fig. 2B, Middle). Therefore, we examined p62 distribution in MEFs co-treated with MG132 and MeHg. As reported previously (Takanezawa et al., 2017), exposure to MeHg increased the number of p62 puncta in MEFs (Fig. 2C, Lower Left). Similarly, MG132-treated cells exhibited a marked increase in the number of p62 puncta compared to that in MeHg-treated cells, with widespread distribution in cells (Fig. 2C, Upper Right). In MEFs co-treated with MG132 and MeHg, p62 puncta were mostly localized close to the nucleus as large puncta (Fig. 2C, Lower Right). Reportedly, p62 overexpression can enhance protein aggregation and has a protective effect on cell survival (Bjørkøy et al., 2005; Salminen et al., 2012). Thus, the formation of large p62 puncta may mitigate proteotoxicity in cells co-treated with MG132 and MeHg. However, it remains to be determined whether p62 granules formed by MeHg and/or MG132 can co-localize with proteasomes. Further studies to examine p62 granules formed by MeHg co-localization with proteasome will help clarify the mechanisms.

Inhibition of proteasome or autophagy hinders the degradation of cytoplasmic proteins, resulting in cellular accumulation as ubiquitinated proteins (Adams, 2003; Korolchuk et al., 2009), and p62 plays a crucial role in targeting the two proteolytic pathways and contributes to cell survival (Cohen-Kaplan et al., 2016; Shin et al., 2020). We examined whether p62 can impact the viability of MEFs following co-treatment with MeHg and a proteasome or autophagy inhibitor (MG132 or CQ, respectively (Fig. 3). CCK-8 assays showed that co-treatment with MeHg and MG132 or CQ significantly reduced p62KO MEF cell viability compared to that of wild-type cells, suggesting that p62 is vital in mitigating MeHg toxicity via both proteasomes and autophagy.
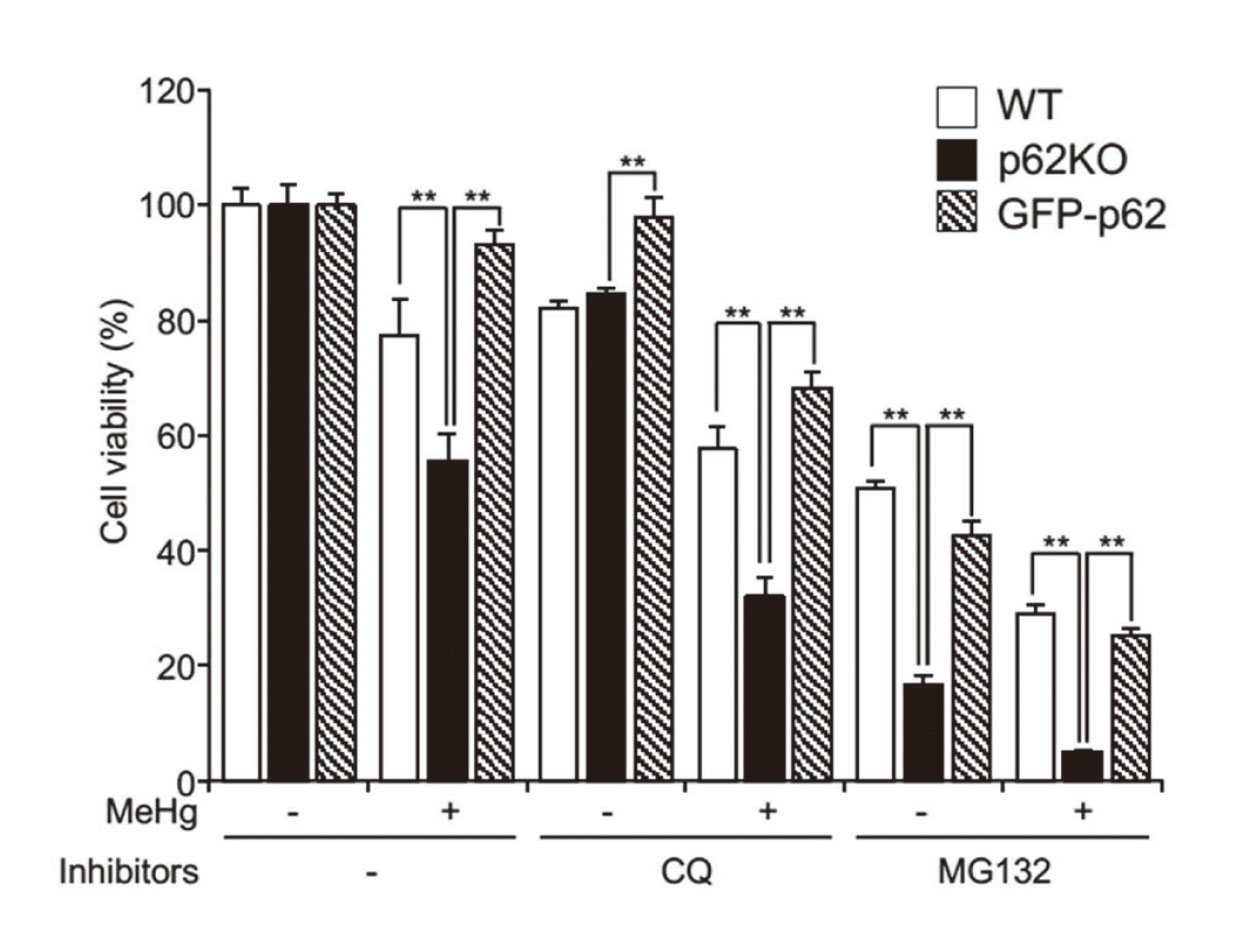
To the best of our knowledge, for the first time, we demonstrated that proteasome inhibitors enhance MeHg toxicity in mammalian cells independent of elevated intracellular Hg concentrations. Further, our results suggest that p62 may play an important role in transporting MeHg-induced ubiquitinated proteins to proteasomes in addition to autophagosomes. These results suggest that the p62–proteasome axis serves as a protective pathway to attenuate MeHg toxicity. In addition to the role of p62 in autophagy (Takanezawa et al., 2016, 2017), we postulate that p62 protects cells from proteasome inhibitors and MeHg-induced toxicity by removing ubiquitinated proteins via proteasome. On the other hand, p62KO cells failed to reduce intracellular MeHg concentrations compared to wild-type cells (Supplementary Fig. 3), suggesting that p62 contributes to the reduction of intracellular MeHg levels. These results collectively indicate that p62 is an important player in not only removing ubiquitinated proteins but also reducing intracellular MeHg levels. Consequently, p62 may attenuate cell death signaling pathways, such as endoplasmic reticulum stress, following exposure to a proteasome inhibitor and MeHg (Tominaga et al., 2017; Takanezawa et al., 2021). Hence, we propose that both p62–proteasome and p62–autophagy axes warrant further evaluation as potential mechanisms for attenuating MeHg toxicity.
ACKNOWLEDGMENTS
We thank Dr. Masaaki Komatsu (Faculty of Medicine, Juntendo University) and Dr. Toru Yanagawa (Division of Medicine, University of Tsukuba) for providing p62KO MEFs. We thank R. Harada, T. Sugimoto, K. Sakai, K. Uchibori, R. Adachi, and L. Yamamoto for their technical assistance. We would like to thank Editage (www.editage.com) for English language editing. This work was partly supported by Grants-in-Aid for Scientific Research C (Grant Number 22K12392) and the Study of the Health Effects of Heavy Metals organized by the Ministry of Environment, Japan.
Conflict of interest
The authors declare that there is no conflict of interest.
REFERENCES
- Adams, J. (2003): The proteasome: structure, function, and role in the cell. Cancer Treat. Rev., 29 (Suppl 1), 3-9.
- Amm, I., Sommer, T. and Wolf, D.H. (2014): Protein quality control and elimination of protein waste: the role of the ubiquitin-proteasome system. Biochim. Biophys. Acta, 1843, 182-196.
- Auger, N., Kofman, O., Kosatsky, T. and Armstrong, B. (2005): Low-level methylmercury exposure as a risk factor for neurologic abnormalities in adults. Neurotoxicology, 26, 149-157.
- Bjørkøy, G., Lamark, T., Brech, A., Outzen, H., Perander, M., Øvervatn, A., Stenmark, H. and Johansen, T. (2005): p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol., 171, 603-614.
- Clarkson, T.W. and Magos, L. (2006): The toxicology of mercury and its chemical compounds. Crit. Rev. Toxicol., 36, 609-662.
- Cohen-Kaplan, V., Livneh, I., Avni, N., Fabre, B., Ziv, T., Kwon, Y.T. and Ciechanover, A. (2016): p62- and ubiquitin-dependent stress-induced autophagy of the mammalian 26S proteasome. Proc. Natl. Acad. Sci. USA, 113, E7490-E7499.
- Hiraoka, H., Nakahara, K., Kaneko, Y., Akiyama, S., Okuda, K., Iwawaki, T., Fujimura, M., Kumagai, Y., Takasugi, N. and Uehara, T. (2017): Modulation of unfolded protein response by methylmercury. Biol. Pharm. Bull., 40, 1595-1598.
- Hwang, G.-W., Furuchi, T. and Naganuma, A. (2002): A ubiquitin-proteasome system is responsible for the protection of yeast and human cells against methylmercury. FASEB J., 16, 709-711.
- Hwang, G.-W., Sasaki, D. and Naganuma, A. (2005): Overexpression of Rad23 confers resistance to methylmercury in saccharomyces cerevisiae via inhibition of the degradation of ubiquitinated proteins. Mol. Pharmacol., 68, 1074-1078.
- Karagas, M.R., Choi, A.L., Oken, E., Horvat, M., Schoeny, R., Kamai, E., Cowell, W., Grandjean, P. and Korrick, S. (2012): Evidence on the human health effects of low-level methylmercury exposure. Environ. Health Perspect., 120, 799-806.
- Ke, T., Gonçalves, F.M., Gonçalves, C.L., Dos Santos, A.A., Rocha, J.B., Farina, M., Skalny, A., Tsatsakis, A., Bowman, A.B. and Aschner, M. (2019): Post-translational modifications in MeHg-induced neurotoxicity. Biochim. Biophys. Acta Mol. Basis Dis., 1865, 2068-2081.
- Komatsu, M., Kurokawa, H., Waguri, S., Taguchi, K., Kobayashi, A., Ichimura, Y., Sou, Y.S., Ueno, I., Sakamoto, A., Tong, K.I., Kim, M., Nishito, Y., Iemura, S., Natsume, T., Ueno, T., Kominami, E., Motohashi, H., Tanaka, K. and Yamamoto, M. (2010): The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol., 12, 213-223.
- Korolchuk, V.I., Mansilla, A., Menzies, F.M. and Rubinsztein, D.C. (2009): Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell, 33, 517-527.
- Kumagai, Y., Kanda, H., Shinkai, Y. and Toyama, T. (2013): The role of the Keap1/Nrf2 pathway in the cellular response to methylmercury. Oxid. Med. Cell. Longev., 2013, 848279.
- Liu, W.J., Ye, L., Huang, W.F., Guo, L.J., Xu, Z.G., Wu, H.L., Yang, C. and Liu, H.F. (2016): p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett., 21, 29.
- Salminen, A., Kaarniranta, K., Haapasalo, A., Hiltunen, M., Soininen, H. and Alafuzoff, I. (2012): Emerging role of p62/sequestosome-1 in the pathogenesis of Alzheimer’s disease. Prog. Neurobiol., 96, 87-95.
- Seibenhener, M.L., Babu, J.R., Geetha, T., Wong, H.C., Krishna, N.R. and Wooten, M.W. (2004): Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell. Biol., 24, 8055-8068.
- Shin, W.H., Park, J.H. and Chung, K.C. (2020): The central regulator p62 between ubiquitin proteasome system and autophagy and its role in the mitophagy and Parkinson’s disease. BMB Rep., 53, 56-63.
- Takanezawa, Y., Nakamura, R., Harada, R., Sone, Y., Uraguchi, S. and Kiyono, M. (2017): Sequestosome1/p62 protects mouse embryonic fibroblasts against low-dose methylercury-induced cytotoxicity and is involved in clearance of ubiquitinated proteins. Sci. Rep., 7, 16735.
- Takanezawa, Y., Nakamura, R., Matsuda, H., Yagi, T., Egawa, Z., Sone, Y., Uraguchi, S., Adachi, T. and Kiyono, M. (2019a): Intracellular demethylation of methylmercury to inorganic mercury by organomercurial lyase (MerB) strengthens cytotoxicity. Toxicol. Sci., 170, 438-451.
- Takanezawa, Y., Nakamura, R., Sone, Y., Uraguchi, S. and Kiyono, M. (2019b): An autophagy deficiency promotes methylmercury-induced multinuclear cell formation. Biochem. Biophys. Res. Commun., 511, 460-467.
- Takanezawa, Y., Nakamura, R., Sone, Y., Uraguchi, S. and Kiyono, M. (2016): Atg5-dependent autophagy plays a protective role against methylmercury-induced cytotoxicity. Toxicol. Lett., 262, 135-141.
- Takanezawa, Y., Nakamura, R., Sugimoto, T., Ohshiro, Y., Uraguchi, S. and Kiyono, M. (2021): p62/sequestosome 1 attenuates methylmercury-induced endoplasmic reticulum stress in mouse embryonic fibroblasts. Toxicol. Lett., 353, 93-99.
- Tominaga, T., Goto, M., Onoue, T., Mizoguchi, A., Sugiyama, M., Tsunekawa, T., Hagiwara, D., Morishita, Y., Ito, Y., Iwama, S., Suga, H., Banno, R. and Arima, H. (2017): Sequestosome 1 (SQSTM1/p62) maintains protein folding capacity under endoplasmic reticulum stress in mouse hypothalamic organotypic culture. Neurosci. Lett., 656, 103-107.
- Unoki, T., Akiyama, M., Kumagai, Y., Gonçalves, F.M., Farina, M., da Rocha, J.B. and Aschner, M. (2018): Molecular pathways associated with methylmercury-induced Nrf2 modulation. Front. Genet., 9, 373.
- Usuki, F., Fujimura, M. and Yamashita, A. (2017): Endoplasmic reticulum stress preconditioning modifies intracellular mercury content by upregulating membrane transporters. Sci. Rep., 7, 12390.
- Wang, L., Jiang, H., Yin, Z., Aschner, M. and Cai, J. (2009): Methylmercury toxicity and Nrf2-dependent detoxification in astrocytes. Toxicol. Sci., 107, 135-143.
- Yokoo, E.M., Valente, J.G., Grattan, L., Schmidt, S.L., Platt, I. and Silbergeld, E.K. (2003): Low level methylmercury exposure affects neuropsychological function in adults. Environ. Health, 2, 8.