Original Article
Neuro-protective effects of n-butylphthalide on carbon monoxide poisoning rats by modulating IL-2, AKT and BCL-2
2023 Volume 48 Issue 9 Pages 495-505
Details
2023 Volume 48 Issue 9 Pages 495-505
Acute carbon monoxide poisoning (CO-poisoning) causes neurotoxicity by inducing necrosis, apoptosis, lipid peroxidation, and oxidative stress. DL-3-n-butylphthalide (NBP) is a synthetic compound originally extracted from the seeds of Chinese celery and based on pure l-3-n-butylphthalide. In ischemia/reperfusion, it exerts neuroprotective effects through its anti-apoptotic, anti-necrotic and antioxidant properties, and activation of pro-survival pathways. Our study performed bioinformatic analysis to identify the differential expression genes. CO-poisoning patients’ blood was collected to confirm the findings. Male rats were exposed to CO 3000 ppm for 40 min, and NBP (100 mg/kg/day) was continuously injected intraperitoneally immediately after poisoning and for the next 15 days. After NBP treatment, the rats were evaluated by Morris water maze test. At the end of experiments, blood and brain tissues of the rats were collected to evaluate the expression levels of IL-2, AKT and BCL-2. We found that IL-2 was elevated in CO-poisoning patients and animal models. Brain tissue damage in CO-poisoning rats was significantly alleviated after NBP treatment. Furthermore, NBP increased the expression of IL-2, AKT and BCL-2 in rat CO-poisoning model. NBP showed neuroprotective action by increasing IL-2, AKT, and BCL-2 expressions.
Carbon monoxide (CO) is a poisonous gas produced by incomplete burning of fossil fuels (Ashcroft et al., 2019). A large number of patients die from monoxide carbon poisoning (CO-poisoning) each year (Rose et al., 2017). Severe acute CO-poisoning manifests as cognitive dysfunction and progresses rapidly due to progressive brain injury and edema. CO-poisoning patients suffer from long period of neurocognitive dysfunction accompanied with brain damage, which manifests as cognitive dysfunction, memory impairment, anxiety, depression, motor, and vestibular deficits. Although symptoms could be relieved with months or even as long as 1 year, studies have shown that after 6 years of CO-poisoning, the incidence of cognitive deficits in patients is 19% and the incidence of neurologic deficits is 37%. The long-term mortality rate of survivors of acute CO-poisoning is almost twice that of the standard population (Rose et al., 2017).
The current treatment for CO-poisoning is pure normobaric oxygen or hyperbaric oxygen (HBO2) (2.5–3 atmospheres). HBO2 has been shown to improve the effect of mitochondrial dysfunction and inflammation induced by CO-poisoning. Although HBO2 is obviously effective for CO-poisoning patients, there are still about 10,000 to 20,000 survivors each year who subsequently suffer from permanent neurocognitive and affective sequelae, which indicates the need for research on new therapies. Besides, some methods for managing the downstream effects of CO-poisoning, which are associated with the induction of reperfusion injury and oxidation and inflammation signaling pathways, and CO removal methods have also been investigated. These treatments for inflammation and oxidative stress caused by CO-poisoning may be effective. Therefore, in addition to effective public awareness and public safety precautions, the clinical need for better therapy for CO-poisoning has not yet been met.
DL-3-n-butylphthalide (NBP) is firstly extracted from the seeds of Chinese celery, and then synthesized based on pure l-3-n-butylphthalide (Zhi et al., 2020). It has been approved by National Medical Products Administration (NMPA) of China for clinical treatment of stroke patients. Protective effects of NBP on ischemic brain damages have been indicated. It can reduce infarct volume, reduce neurovascular damage, and improve cognitive functions and neurological outcomes. Recently, it was discovered that NBP has a neuroprotective effect on brain injury after acute CO-poisoning (Xiang et al., 2017). Therefore, we implemented this study to explore the protect effects of NBP on cognitive dysfunction in CO-poisoning rat models.
In this study, we found that IL-2 may be involved in the progression of CO-poisoning. NBP may exhibit neuroprotective effects in CO-poisoning by increasing the expression of IL-2, AKT and BCL-2. Therefore, this study may provide further basis for the application of NBP to brain damage induced by CO-poisoning.
Microarray of GSE94780 was obtained from GEO database (https://www.ncbi.nlm.nih.gov/gds/) (Hara et al., 2017). The heatmap and volcano figures were analyzed and generated by R program (version R 4.1.2).
AgentsAntibodies against BCL-2 (ab196495), AKT (phospho T308, ab8933) and β-actin (ab8227) as well as secondary antibodies were purchased from Abcam (USA). DL-3-n-butylphthalide (NBP) was obtained from Shijiazhuang Pharmaceutical Group Co., Ltd. (Hebei, China).
Patient recruitmentThe purpose and design of this study were reviewed and approved by the Committee of Human Ethics of the First People's Hospital of Chenzhou. All the blood of CO-poisoning patients (n = 10), which administrated at emergency within 1 hr, and healthy cohort (n = 10) were collected with written consent. This work was conducted from November 2019 to January 2021.
Animals and NBP treatmentMale Wistar rats (8 to 10 weeks old, 200 to 250 g) were ordered from Huafukang Co. (Beijing, China), and each cage was assigned with 3 rats randomly. The rats were kept under specific pathogen-free condition with a 12 hr light/dark cycle, and free access to standard food and water. The room temperature was 21 to 24°C, and the relative humidity was 40–60%. The rats in the plastic chamber were exposed to room air alone (control, n = 6) for 40 min or 3000 ppm CO (n = 12, divided into two groups treated with or without NBP randomly) (Hara et al., 2017). The concentration of CO was controlled using gas flow regulator (YQAr-731L, Ronghua, Shanghai, China) and adjusted to 3000 ppm, the flow rate was monitored with a CO (GASTiger3000-CO, Wandi, Shenzhen, China) or O2 (EDW-100-O2, Wandi, Shenzhen, China) monitor. After CO-poisoning rat model was established, NBP (100 mg/kg) or saline was intraperitoneally administered within 5 min at first day and daily for 15 days as described before (Zhao et al., 2017). All rat behavioral experiments were approved by the Committee of Animal Care and Experiment of Southern Medical University, China.
Morris water maze testMorris water maze test was performed following the previous method (Morris, 1984; Xiong et al., 2017) on days 11-15th of NBP administration. The test consisted of a circular pool with a diameter of 160 cm, filled with opaque water (containing white food coloring) at 22 ± 1°C. The pool was separated as four quadrants. A hidden platform was placed 2 cm below the water in one quadrant, and different visual cues were fixed on the pool wall. Walls with black cloth were around the water maze to create a covered area of 4 × 6 m. In the place navigation test, the rats were randomly placed into the water from one of the quadrants and performed four experiments a day for five consecutive days. The time spent to get to the platform (escape latency) was recorded. If a rat did not find the platform within 120 sec, the trainer would guide the rat to reach the platform and rest on the platform for 30 sec. The time was recorded as 120 sec. On the sixth day, the underwater platform was removed, and the spatial probe test was carried out. The rat was placed into the water from the opposite quadrant where the platform had been placed and allowed to swim for 120 sec. The ratio of the time that rats spent in the target quadrant was recorded.
Carboxyhemoglobin level assessmentTo confirm CO-poisoning, blood samples were taken from the tail of rats after being exposed to CO, the blood was kept, and carboxyhemoglobin (COHb) levels were measured by spectrophotometer (Shimadzu UV-1800, Japan).
Blood IL-2 measurementWhole blood samples (1 mL) from CO-poisoning patients or animal models were obtained. The human and rat IL-2 ELISA (enzyme-linked immunosorbent assay, Solarbio, Beijing, China) kits were used to measure the level of IL-2 in the supernatant according to its standard curve. All samples were tested in duplicate, the results showed low variability between replicates, and all were within the measured standard curve.
Sample preparationsAt the end of the behavior experiment, the rats were euthanized with sodium pentobarbital (50 mg/kg, ip). The brain tissues were removed after perfusion with cold saline. The samples were weighed and dissected on ice. Half of the samples were immersed in RNA-Later stabilization solution (Ambion, CA, USA) at 4°C overnight, stored at -80°C for RNA extraction, and the other half was stored in liquid nitrogen for western blotting or fixed in formalin for paraffin embedding.
RT-qPCRRat brain tissue was lysed and homogenized on ice. TRIzol reagent (Invitrogen, USA) was applied to extract total RNA from rat brain tissue. RevertAid First Strand cDNA Synthesis kit (Invitrogen, USA) was used to synthesize cDNA by reverse transcription using total RNA as template. SYBR Premix Ex Taq (Takara Bio, Japan) were used for qPCR amplification, using the following thermal cycling conditions: initial denaturation at 95°C for 30 sec, denaturation at 95°C for 15 sec, annealing and extension at 60°C for 1 min, followed by 35 cycles, IL-2 mRNA expression was qualified by the 2-ΔΔCt method, and β-actin was used as an internal standard for normalization. The primers are as follows: IL-2 F: 5'- GGGATCTGAAACAACATTCATGTG -3'; R: 5'- AGTCAGTGTTGAGATGATGCTTTG -3' (Taheri et al., 2021). β-actin F:5'- GGGAAATCGTGCGTGACAT -3'; R: 5'- GGAGCCAGGGCAGTAATCT -3'.
Western blotProteins were extracted from rat brain tissues. Then proteins were separated by electrophoresis in the 10% polyacrylamide gel and transferred to Polyvinylidene fluoride (PVDF) membranes (Merck, MA, USA). Membranes were blocked in 5% fat-free milk for 1 hr at room temperature and incubated with primary antibodies (concertation: BCL-2, 1:1000; AKT, 1:1000; β-actin, 1:5000) at 4°C overnight. The next day, membranes were rinsed three times with TBST and incubated with horseradish peroxidase-conjugated secondary antibody (1:1000) for 1 hr at room temperature. The bolts were detected by enhanced chemiluminescence (ECL) kit (GE Healthcare, UK), then the film was fixed and developed (Solarbio, Beijing, China). The experiments were performed at least three times. Protein expression level was normalized against β-actin and measured by using Image J system (National Institutes of Health, Bethesda, MD, USA).
Hematoxylin and eosin stainingHematoxylin and eosin (HE) staining was conducted according to manufactory’s protocol (Solarbio, Beijing, China). Paraffin-embedded rat brain tissues were cut into 4-μm-thin sections. The slides were stained with hematoxylin for 5 min, subsequently with eosin for 30 sec. Then, the slides were dried and mounted using resin. Histological examinations were assessed by two pathologists. Images were recorded by microscopy (Olympus CX23; Olympus Corporation, Tokyo, Japan).
StatisticsThe data are shown as mean ± SD. SPSS (version 21.0, IBM, USA) was used for statistical analysis. Differences between multiple groups were compared by one-way ANOVA (Dunnett's or Turkey's tests). Differences at p value < 0.05 were considered as statistically significant.
Microarray analysis of GSE94780 was performed by R program. The differential expression genes (DEGs) are shown in heatmap (Fig. 1A) and volcano figure (Fig. 1B). We found that the mRNA expression of IL-2 was significantly upregulated in the CO-poisoning group compared with the control group. Moreover, the GO analysis showed that transcription factor complex, skeletal muscle tissue development, skeletal muscle cell differentiation and skeletal muscle organ development were enriched (Fig. 1C-D).

Bioinformatic analysis identified IL-2 was upregulated in CO-poisoning rat model. A: Heatmap showed IL-2 expression was upregulated in rat model. B: Volcano figure showed IL-2 expression was upregulated in rat model. C: GO analysis showed transcription factor complex was upregulated. D: GO analysis showed skeletal muscle differentiation, development was upregulated.
To confirm the bioinformatic findings that the mRNA expression of IL-2 was upregulated in CO-poisoning model, the level of IL-2 in CO-poisoning patients’ blood was tested. The result showed that blood IL-2 concentration was increased in CO-poisoning patients (n = 10) compared with healthy cohort (n = 10) (259.60 ± 53.92 pg/mL vs. 89.11 ± 25.35 pg/mL) (p < 0.05) (Fig. 2).

IL-2 level was increased in CO-poisoning patients’ blood. Blood IL-2 concentration was increased in CO-poisoning patients (n = 10) compared with healthy cohort (n = 10) (*p < 0.05).
To further investigate whether NBP showed a protective effect on memory, the rat CO-poisoning model was established. CO at 3000 ppm caused over 50% COHb in the blood. Then, the CO-poisoning rats were treated with or without NBP, the Morris water maze test was performed to evaluate the neuroprotective action of NBP subsequently. Compared with the control and CO-poisoning groups, NBP not only reduced the escape latency time (Control vs. CO-poisoning vs. NBP: 60.52 ± 8.03 vs. 90.29 ± 6.44 vs. 84.97 ± 7.32 s, Fig. 3A), but also improved the percentage of time (Control vs. CO-poisoning vs. NBP: 39.01 ± 2.40 vs. 24.74 ± 2.49 vs. 32.00 ± 2.69%, Fig. 3B). In addition, the HE staining of rat brain tissue showed that the brain damage of CO-poisoning rats after NBP treatment was significantly reduced (Fig. 4).
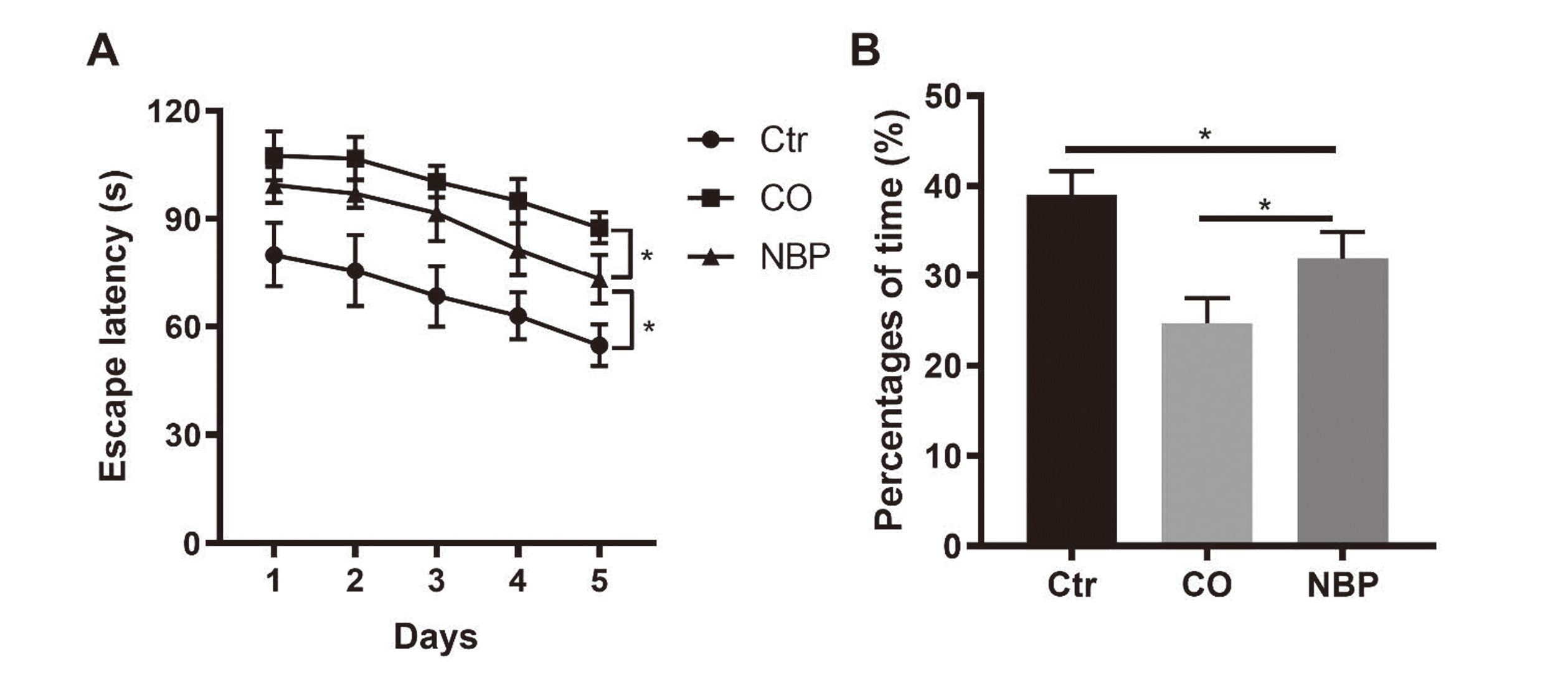
A: Difference in escape latency between NBP group and MCP group B: Difference in latency time between NBP group and MCP group (n = 6) (*p < 0.05).
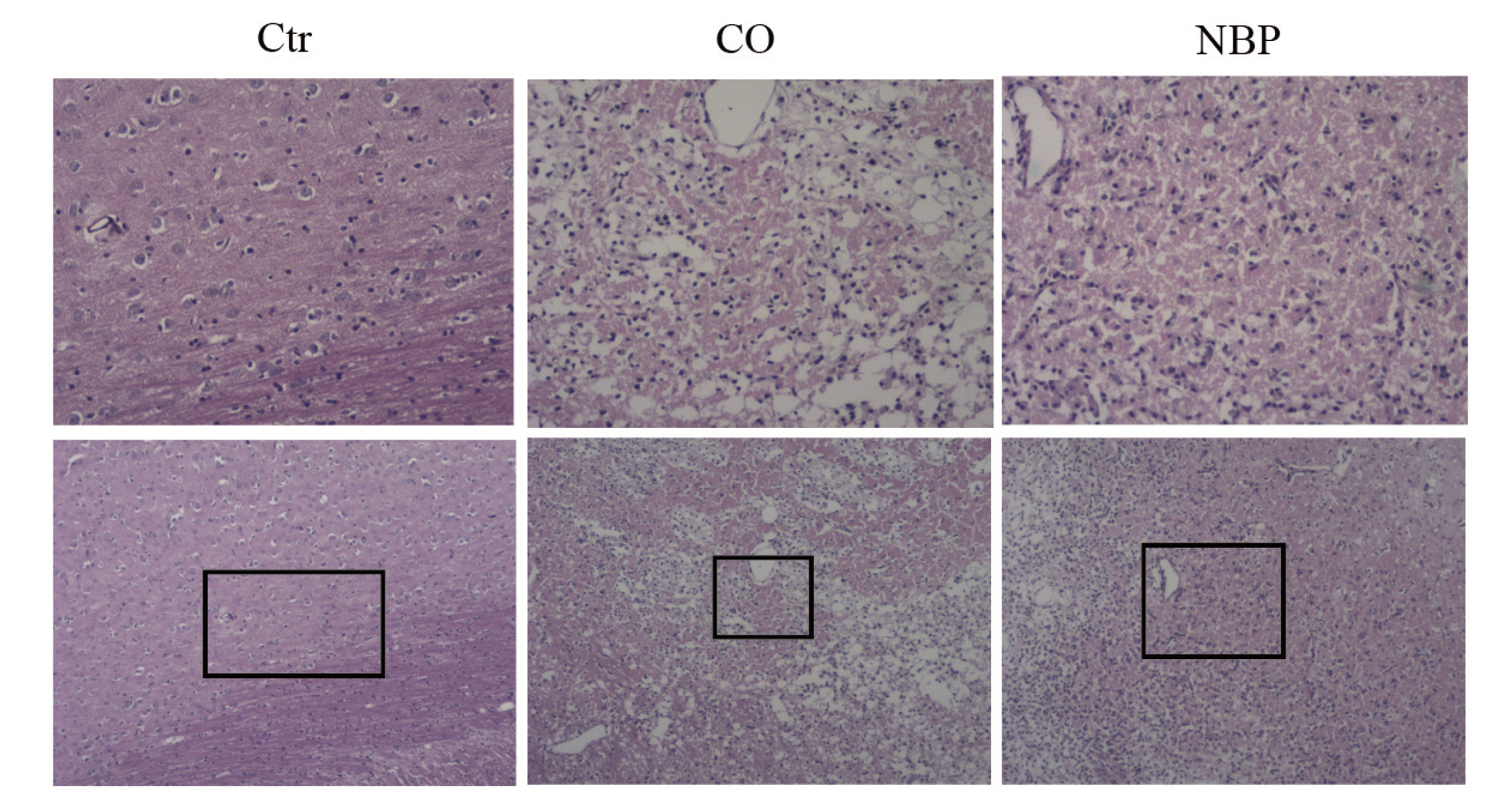
The HE staining of rats brain tissues. The upper panels are the magnificantions (×100) of the lower panels (×40).
To understand the mechanism of the neuroprotective effect of NBP, the mRNA expression of IL-2 in rat brain and the level of IL-2 in rat blood were detected. Our results showed that mRNA level of IL-2 was increased in the CO-poisoning group (Fold change: 1.45 ± 0.12), and was higher in the NBP group (Fold change: 2.62 ± 0.19) (Fig. 5A). Consistently, the level of IL-2 in rat blood was increased in the CO-poisoning group, and was higher in the NBP group as well (Control vs. CO-poisoning vs. NBP: 18.48 ± 2.67 vs. 36.47 ± 5.6 vs. 74.31 ± 15.74 pg/mL, Fig. 5B). Furthermore, western blots showed that the expression of AKT and BCL-2 was increased in the CO-poisoning group, and was higher in the NBP group (Fig. 6 and Supplementary Fig. 1).
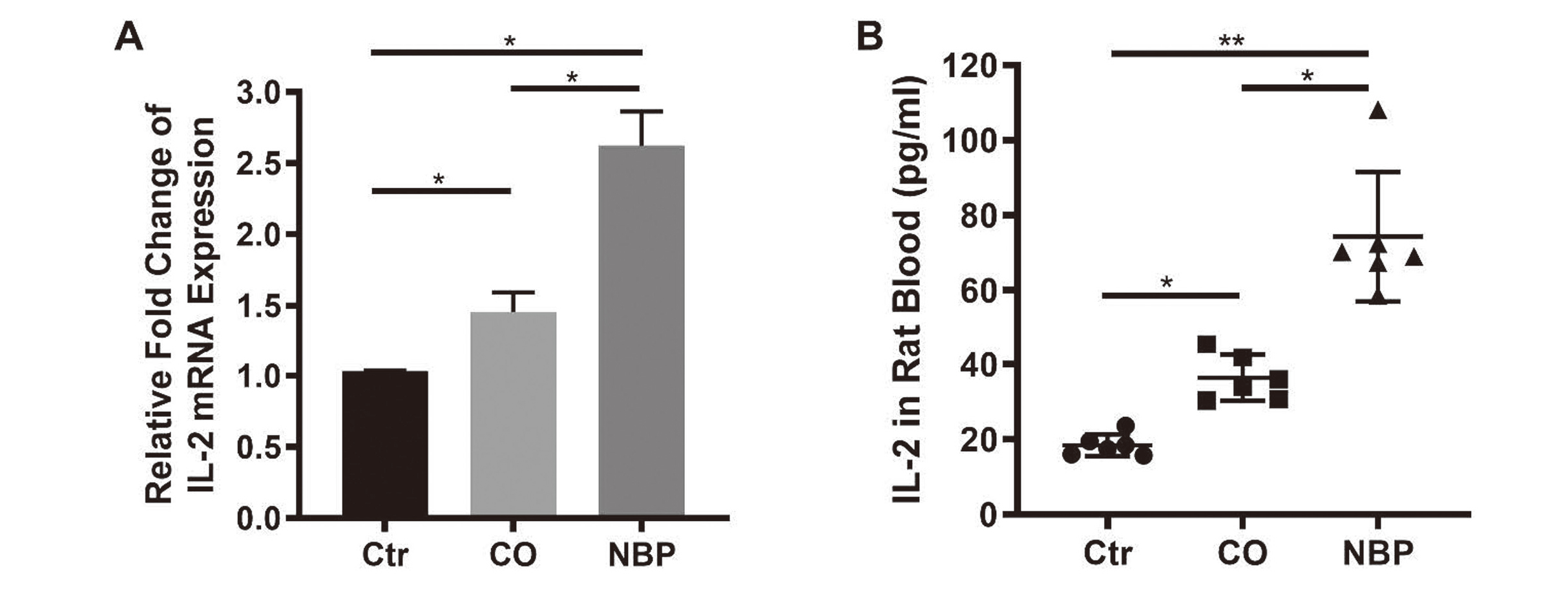
IL-2 level was increased in CO-poisoning rat model and higher in NBP treatment. A: The mRNA expression of IL-2 in rat brain tissues (n = 6); B: The level of IL-2 in rat blood (*p < 0.05, **p < 0.01).
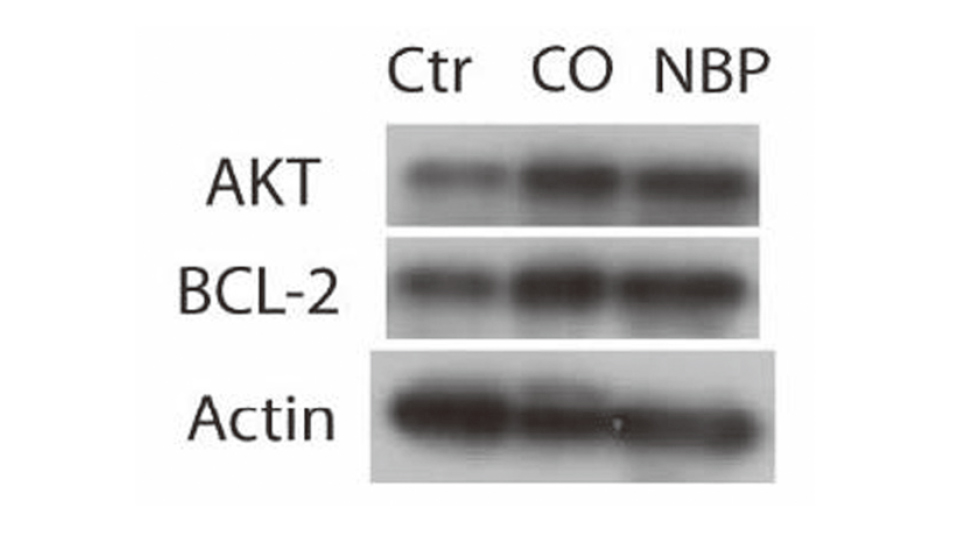
NBP increases AKT and BCL-2 expression in rat CO-poisoning model. The expression of AKT, BCL-2 and beta-actin was detected by western blot.
Acute CO-poisoning has severe consequences on the brain. Previous investigations suggest that NBP is a promising multifaceted drug for the therapy of CO-poisoning (Xiang et al., 2017). However, it is not yet clear what is the mechanism of therapeutic benefits. To solve these problems, a CO-poisoning model was induced in adult male rats. Our present study found that the level of IL-2 was increased in CO-poisoning patients and the rat model. Moreover, NBP showed neuroprotective function in vivo by the potential mechanism that increasing the expression of IL-2, AKT, and BCL-2.
When inhaled in high quantities CO gas enters the bloodstream and binds to haemoglobin (Hb) forming carboxyhaemoglobin (COHb), with a much greater affinity (230 times higher) than oxygen. The COHb results in reduced delivery of oxygen to tissues, which leads to tissue ischemia (Ashcroft et al., 2019). Growing evidence suggests that oxidative stress and secondary reactions in delayed brain injury are crucial to CO toxicity, similar to ischemia-reperfusion injury (Fan et al., 2016).
Tissue hypoxia and reduction of mitochondrial oxidative phosphorylation caused by CO-poisoning lead to ischemic and hypoxic brain damage (Tabrizian et al., 2019). COHb not only hinders the combination of oxygen and Hb but also inhibits the dissociation of oxygen, causing respiratory dysfunction in cell mitochondria and reducing ATP production (Li et al., 2015). CO can also bind to the cytochrome aa3 in the mitochondria of the cell to block the electron transmission in the respiratory chain. In the process of reperfusion, a large number of free radicals are generated (Bagheri et al., 2019). MPO and ROS will attack lipid membranes, degrade unsaturated fatty acids and cause lipid peroxidation, form adducts with myelin basic protein, and trigger lymphocyte reaction and microglial activation (Rose et al., 2017). These inflammatory effects persist for a long time after the initial CO-poisoning and may not be related to the COHb level (Ashcroft et al., 2019). CO-poisoning may cause cognitive dysfunction in survivors through the above-mentioned mechanisms. It is well documented that immune cytokines and cells have different effects on center nervous system (CNS) recovery after injury including CO-poisoning. Inflammatory stimulation exerts neuroprotective, axon-growth-promoting and significant disinhibitory effects (Leibinger et al., 2013). Our study found that the expression of IL-2 was increased in CO-poisoning patients and the rat model.
IL‐2 is known as a growth and survival factor that mediates proliferation of CD4+ T cells and the differentiation of the memory T cell phenotype (Kanagalingam et al., 2019). Several studies have brought evidence that IL-2 plays a protective role in ischemia-reperfusion injury of brain. Early studies using the mouse ischemic brain stroke injury model observed increased expression of IL-2 and a massive accumulation of Treg cells during the chronic ischemic phase, which indicated potentiation of neurological recovery (Ito et al., 2019). In a later study, Zhao and colleagues observed that IL-2 levels were higher in acute ischemic stroke (AIS) patients with favorable outcomes than in those with poor outcomes. In addition, the imbalance between circulating neuroprotective (Th2 and Treg) and neurotoxic (Th1 and Th17) CD4+ T lymphocyte subsets also affects prognosis in AIS. Decreasing circulating Tregs and increasing Th17 have been shown to contribute to poor prognosis in AIS (Zhao et al., 2020). Our findings showed that IL-2 level was increased in CO patients and animal model. These observations imply that the increasing expression of IL-2 can be considered as a compensatory protective response after CNS injury induced by CO poisoning. It is conceivable that NBP treatment improved the neuroprotective effect by further enhancing the expression level of IL-2.
Several lines of evidence suggest that IL-2 showed antiapoptotic and proliferative effects via induction of BCL‐2 and AKT protein kinase. BCL-2 is also known as an essential regulator of sympathetic neurons survival after nerve growth factor deprivation (Kanagalingam et al., 2019; Gómez et al., 1998) and can inhibit oxidant-induced apoptosis. The BCL-2 family includes apoptosis-suppressing genes and apoptosis-promoting genes. The molecules regulate cell apoptosis by forming dimers of different quality and quantity. Studies have shown that IL-2 can induce the expression of BCL-2 (Shinetova et al., 2020), which may be mediated by NF-AT (Gómez et al., 1998). IL-2 can stimulate human peripheral blood lymphocytes to cause the accumulation of BCL-2α and β mRNA (Artyukhov and Tyunina, 2017; Pae et al., 2004). A previous study reported that Th2 cells cultured in high IL‐2 exhibited an increasing of BCL‐2 (anti-apoptosis protein) and decreasing of BIM (pro-apoptosis protein) (Kanagalingam et al., 2019). Our results showed the up regulation of IL-2 and BCL-2 in CO-poisoning rat model, which indicated the protective mechanism was activated.
The link between BCL-2 and AKT is reinforced by reports that resveratrol showed neuroprotective effect in CO-poisoning rats as it decreased necrosis and BAX/BCL-2 ratio and increased AKT expression levels (Tabrizian et al., 2017). In another animal CO-poisoning model treated with hesperidin, AKT protein expression was increased, while the BAX/BCL-2 ratio was significantly decreased (Rezaee et al., 2019). The PI3K/AKT is a classic pro-proliferation and anti-apoptotic signal pathway. It plays a key regulatory role in the process of cerebral ischemia-reperfusion injury. Activating the PI3K/AKT pathway can inhibit brain inflammation and oxidative stress, thereby inhibiting nerve cells apoptosis and maintain normal nerve function (Tabrizian et al., 2017). The up-regulation of AKT transcription inhibits the conformational changes of the pro-apoptotic BAX protein and its translocation to mitochondria, thereby avoiding the destruction of the inner mitochondrial membrane and the activation of caspase-3 (Hashemzaei et al., 2016). In line with these observations, our data demonstrated that increasing of BCL-2 and AKT attenuates the injury from CO.
However, it was of great interest to notice that CO had an antiproliferative effect on CD4+ T cells at the low concentration (200 ppm) and inhibited IL-2 secretion by activated CD4+ T cells (Pae et al., 2004). While the exogenous IL-2 effectively reversed the antiproliferative effects of CO in T cell culture, CO-releasing compound RuCO suppressed the proliferative response and the secretion of IL-2 by CD4+ T cells as well, but not in the presence of the CO scavenger Hb.
A previous study reported on the anti-cerebral ischemic effects of NBP, which can reduce cerebral edema as well (Taheri et al., 2021). NBP can increase blood flow in ischemic areas of the brain and improve brain energy metabolism, thereby inhibit nerve cell death, and improve the memory function of specific nerve cells, finally effectively reversing or improving cognitive dysfunction (Min et al., 2014; Wang et al., 2019). In addition, it has anti-apoptotic, anti-inflammatory, anti-platelet, and anti-thrombosis functions (Zheng et al., 2017). However, the specific molecular mechanism of NBP's action is still unclear.
Unexpectedly, we didn’t find the increasing of BCL-2 and AKT in Fig. 1. But we noticed some of the differential expression genes correlating with gene transcription and cell survival, such as BCL6B, GRAP2, NMU, NMUR2, ATF3, Fos and NTF3 et al., may play important role in neural recovery.
BCL6B (B-cell lymphoma 6B transcription repressor) was downregulated as shown in Fig. 1. BCL6B acts as a sequence-specific transcriptional repressor in association with BCL6. Several lines of evidence suggest that BCL6 represses the transcription of genes involved in DNA damage and inflammatory response, which contributes to the recovery of neural system damage. Wei et al. reported that inhibition of BCL6 may attenuate oxidative stress-induced neuronal damage (Wei et al., 2021). Additionally, it is reported that inflammatory stimulation exerts neuroprotective effect after neural injury (Leibinger et al., 2013). Chang et al. found that knockdown of BCL6 prevents the differentiation of unprimed human T cells into T suppressor (Ts) cells, whereas overexpression of BCL6 converts CD8+ T cells into Ts cells. Ts cells inhibit CD4 and CD8 T cell effector function (Chang et al., 2010). Moreover, we found that the expression of GRAP2 was increased. GRAP2 is related with leukocyte-specific protein-tyrosine kinase signaling pathway, which involved in T cell activation (Ma et al., 2001).
We found that the expression of NMU (neuromedin U) and NMUR2 (neuromedin U type 2 receptor) was upregulated. The neuromedin encoded by NMU is a member of neuropeptides. NMUR2 is a G-protein coupled receptor for NMU (Yamamoto et al., 2011), which was exclusively expressed in the CNS (Peng et al., 2019). NMU and its receptors play roles in pain, stress response, energy homeostasis, and immune-mediated inflammatory diseases. It is reported that knockdown of NMUR2 alleviates bone cancer pain by inactivation of the PKC/ERK and PI3K/AKT signal pathways (Johnson et al., 2004). NMU-evoked cytokine release required activation of MEK, and PI3K pathways in T cells. Moreover, the expression of DUSP1 was found decreased, which negatively regulate MEK downstream pathway by dephosphorylating MAP kinase MAPK1/ERK2. We also found PIK3C2G was slightly upregulated (data not shown) by analyzing GEO dataset. The protein encoded by this gene belongs to the PI3K family. Considering the CO-poisoning rat models (GSE94780) were established within 40 min and the samples were collected less than 12 hr, we suggest that the increasing of BCL-2 and AKT may be late events of CO-poisoning.
In addition, we found that ATF3 was downregulated. ATF3 encodes a protein belonging to the cAMP family, repressing transcription from promoters with ATF sites. Hara et al. reported that severe CO-poisoning (3000 ppm) caused an increase in cAMP (Hara et al., 2011). We inferred that increasing of cAMP induced by CO poisoning may accompanied with decreasing of ATF3.
The expression of FOS was downregulated as well. The proteins coded by FOS are subunits of activating protein 1 (AP-1) transcription factor. c-Fos is the main constitution of AP-1 transcription complex. Several papers demonstrated that c-Fos plays a critical role in neuronal apoptosis and inflammation (Chen et al., 2015). Up-regulation of c-Fos associated with neuronal apoptosis (Yang et al., 2022). Additional, c-Fos controls light-induced apoptosis of retinal photoreceptors (Wenzel et al., 2000).
The expression of NTF3 was found to be decreased too. The protein NT3 encoded by NTF3 is a member of the neurotrophin family, which controls survival and differentiation of neurons. It has been shown NT3 may be involved in the maintenance and development of neurons, and function as both nerve growth and brain-derived neurotrophic factor. Our finding was similar with the research conducted by Müller, who reported that NT-3 levels remained low in elderly stroke patients after admission to hospital and the following week of observation (Müller et al., 2022). Moreover, NT3 can improve patients’ sensorimotor function after ischemic stroke (Duricki et al., 2019).
Furthermore, it was found that NT3 expression is developmentally regulated in skeletal muscle and may modulate the number of Schwann cells at neuromuscular synapses (Hess et al., 2007). The published evidence indicated that the Fox, MAPK, and PI3K/AKT signaling pathways are closely related to skeletal muscle development (Dou et al., 2023; Sakuma and Yamaguchi, 2011). These findings are consistent with our data, which may explain why GO analysis showed skeletal muscle development was enriched.
In summary, these data from this study demonstrate that some of the DEGs correlating with gene transcription and cell survival contributing to the recovery beneficial effects after CO-poisoning. Another evidence supporting our hypothesis that CO-poisoning may trigger defense mechanism such as elevated IL-2 to reduce the apoptosis was that stress granule (SG) was enriched by GO analysis (shown in Fig. 1). Once the ischemic insult begins, the faster assembly of SG may underlie the higher rates of neuronal survival by protecting housekeeping mRNAs and/or blocking harmful mRNAs (Aramburu-Núñez et al., 2022). BTG4 gene was found upregulated in Fig. 1A. The encoded protein can bridge CNOT7, a catalytic subunit of the CCR4-NOT complex, to EIF4E. While EIF4E is related to the formation of SG. Moreover, it has been reported that hypoxia affects the expression of housekeeping gene in cells. However, CO-induced hypoxia in rats has little or no effects on the expression of “housekeeping gene” β-actin.
Taking all these observations and previous studies together, we suggest that, by and large, the elevation of IL-2 in CO-poisoning patients and animal model could be considered as a compensatory protective response after CNS injury. IL-2 showed antiapoptotic and proliferative effects via induction of AKT and BCL‐2. While NBP treatment improved the neuroprotective effect by further enhancing the expression level of IL-2, AKT and BCL-2.
In conclusion, our study indicates that NBP plays as a protective role in CO-poisoning-induced brain damage by activating IL-2/AKT/BCL-2.
Funding SourcesThis study was supported by Grant 2020XJ127 (Research Grant of Xiangnan University), 20201055 (Research program of Health Committee of Hunan province), and YXJL-2018-0296-0037 (Medical Awards Foundation of Beijing).
Conflict of interestThe authors declare that there is no conflict of interest.