Abstract
A representative surfactant, benzalkonium chloride (BAC) is used as a disinfectant, but sometimes causes serious side effects, including lung disorders such as interstitial pneumonia. However, its pathogenic mechanisms remain unexplained. In this study, we identified a novel mechanism by which BAC initiates inflammatory responses that may be responsible for its side effects. We firstly investigated whether BAC initiates inflammation, and found that BAC promotes the secretion of the pro-inflammatory cytokine interleukin-1β (IL-1β) but not tumor necrosis factor-α (TNF-α) in macrophages. Interestingly, the IL-1β secretion triggered by the surfactants was completely blocked by the K-ATP channel blocker glibenclamide or the calcium chelating agent 1,2-bis(2-aminophenoxy) ethane-N,N,N',N'-tetraacetic acid (BAPTA)-AM. Moreover, genetic experiments revealed that BAC-dependent IL-1β secretion is mediated by the NLRP3 inflammasome. These results suggest that derangement of ion fluxes associated with the interfacial effects of BAC triggers NLRP3 inflammasome activation and subsequent inflammation. Thus, the NLRP3-dependent mechanisms triggered by BAC may explain the pathogenesis of surfactant-caused adverse effects.
INTRODUCTION
A representative surfactant, BAC is commonly used as a preservative and a disinfectant (Lavorgna et al., 2016). However, there are insufficient data on the safety of BAC, and the examples of BAC-related health hazards such as airway, gastrointestinal mucosal damage, and interstitial pneumonia due to accidental ingestion or inhalation have been reported (Choi et al., 2020; Basketter et al., 2004; Presley et al., 2021; Ling and Highet, 2000). In addition, occupational exposures to BAC cause asthma (Choi et al., 2020). Of note, the Korea Centers for Disease Control and Prevention (KCDC) reported idiopathic lung injury associated with a humidifier disinfectant in Korea, raising great concern about the toxicity of chemical disinfectants (Jeon et al., 2019). This lung injury became known as humidifier disinfectant-related lung injury (HDLI) later. HDLI was a major social problem in the 2000s of Korea, because the number of deaths was 4,306 reported. Since many humidifier disinfectants use BAC as a main ingredient, BAC may be one of the causative substances of HDLI (Song et al., 2022). However, there are few studies on the mechanism of HDLI caused by BAC.
The NLR family pyrin domain containing 3 (NLRP3) inflammasome is a multiprotein complex expressed in immune cells that senses a wide range of stresses such as pathogenic microorganisms, and bioactive substances (Kelley et al., 2019). In particular, our recent studies have demonstrated that chemical compounds, including pharmaceuticals, strongly initiate sterile inflammation by activating the NLRP3 inflammasome (Noguchi et al., 2021; Kagi et al., 2021; Kagi et al., 2022; Sekiguchi et al., 2023; Kagi et al., 2024). The activated NLRP3 inflammasome promotes caspase-1-dependent secretion of the inflammatory cytokines IL-1β and IL-18 (Kelley et al., 2019). Activation of the NLRP3 inflammasome occurs as a two-step process: priming and activation (Kelley et al., 2019). The priming means that the expression of NLRP3 and pro-IL-1β protein is transcriptionally upregulated by activation of the pattern-recognition receptors such as Toll-like receptors (TLRs). In the activation process, NLRP3 is activated by cellular stresses, such as mitochondrial damage, lysosomal destruction, and potassium efflux, and then forms the NLRP3 inflammasome together with caspase-1 and apoptosis-associated speck-like protein containing CARD (ASC). Excessive and prolonged activation of the NLRP3 inflammasome is known to be involved in the development of various inflammatory diseases, including systemic lupus erythematosus, psoriasis, leukoplakia, and atopic dermatitis. In the present study, we examined the potential role of the inflammasome in BAC-induced inflammation, and found that BAC promotes the activation of the NLRP3 inflammasome through cathepsin B-dependent mechanisms, which may explain why BAC causes HDLI.
MATERIALS AND METHODS
Cell culture
THP-1 cells from Japanese Collection of Research Bioresources Cell Bank were cultured in RPMI 1640 containing 10% heat-inactivated FBS, 1% penicillin-streptomycin solution, and Plasmocin at 37°C under a 5% CO2 atmosphere (Kagi et al., 2024). For experiments, THP-1 cells were differentiated for 3 hr with 100 nM PMA on the day before stimulation. Bone marrow-derived macrophages (BMDMs) were isolated from mouse femurs in sterile RPMI 1640 and were cultured in RPMI 1640 containing 10 ng/mL M-CSF (Sino Biological, Kanagawa, Japan) and 10% heat-inactivated FBS and 1% penicillin-streptomycin solution (Noguchi et al., 2021).
Generation of knockout cells
NLRP3, ASC, and caspase-1 knockout (KO) cells were generated using the CRISPR/Cas9 system as previously described (Noguchi et al., 2021). Guide RNAs were designed to target a region in the exon 1 of NLRP3 gene (5ʹ-CTGCAAGCTGGCCAGGTACC-3ʹ), the exon 1 of ASC gene (5ʹ-CAGCACGTTAGCGGTGAGCT-3ʹ), and the exon 2 of caspase-1 gene (5ʹ-AAGCTGTTTATCCGTTCCAT-3ʹ) using CRISPRdirect. Guide RNA-encoding oligonucleotide was cloned into lentiCRISPRv2 plasmid (Addgene), and KO cells were established. To determine the mutations of each gene in cloned cells, we analyzed the genomic sequence around the target region by PCR-direct sequencing using extracted DNA from each clone as a template and the following primers: 5ʹ-AGTGTGGACCGAAGCCTAAG-3ʹ and 5ʹ-TTCTCCTCCCCATTGAAGTC-3ʹ for NLRP3; 5ʹ-TTGGACCTCACCGACAAG-3ʹ and 5ʹ-GCAGCTTTGTTTAGGGGTAGG-3ʹ for ASC; 5ʹ-TACCCAACTGTGAGGAGGGG-3ʹ and 5ʹ-TGGCCCTGAAGCCAGAAATAA-3ʹ for caspase-1.
Reagents and antibodies
All reagents were obtained from commercial sources: Ultrapure LPS (Invivogen, San Diego, CA, USA), PMA and nigericin (Santa Cruz, Dallas, TX, USA), Glyburide (Sigma, St. Louis, MO, USA), MCC950 (Cayman, Ann Arbor, MI, USA), Benzalkonium Chloride (BAC), Ca2+ free medium, and potassium chloride (KCl), SDS, saponin, and Triton X-100 (Nacalai Tesque, Kyoto, Japan), BAPTA-AM (Dojindo, Kumamoto, Japan), E64d and CA-074 Me (Peptide Institute, Osaka, Japan), and alum (Thermo Fisher Scientific, Tokyo, Japan). The Abs used were against NLRP3 (AG-20B-0014) (Adipogen, San Diego, CA, USA); IL-1β (#D3A3Z, #12242), p-JNK (#9251), JNK (#9252), and cathepsin B (#31718) (Cell Signaling, Danvers, MA, USA); ASC (cat# D086-3) (MBL, Nagoya, Japan); and caspase-1 (M-20, A-19), ASC (B-3),β-actin (C4), LAMP2 (H4B4), galectin-3 (M3/38) (Santa Cruz).
Immunoblotting
Proteins from cell culture supernatants were extracted as described previously (Noguchi et al., 2018). Cells were lysed in ice-cold lysis buffer containing 20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 10 mM EDTA, 1% Triton X-100, 1% Sodium deoxycholate, and 1% protease and phosphatase inhibitor mixtures (Nacalai Tesque). Samples were resolved by SDS-PAGE and analyzed as described previously (Tsuchida et al., 2020).
ELISA
Concentrations of IL-1β, TNF-α and IL-6 in cell culture supernatants were measured by specific ELISA kits (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Fluo 4 assay
Ca2+ levels in cytosol were quantified by Fluo 4-AM (Dojindo) according to the manufacturer’s instructions.
Quantitative real time-PCR
Total RNA was extracted from BMDMs with Sepasol RNA I Super (#09379–55, Nacalai Tesque) and reverse transcribed into cDNA with High-Capacity cDNA Reverse Transcription Kit (#4368813, Applied Biosystems, Waltham, MA, USA). Relative mRNA levels of target genes were determined by real-time quantitative fluorescence PCR (qRT-PCR) using Luna Universal qPCR Master Mix (#M3003E, New England Biolabs, Ipswich, MA, USA) according to the manufacturer's protocol (Sekiguchi et al., 2019). Primers used for qRT-PCR: 5’-GAAATGCCACCTTTTGACAGTG-3’ and 5’-CTGGATGCTCTCATCAGGACA-3’ for mouse IL-1β, 5’-ACTTCGGGGTGATCGGTCCCC-3’ and 5’-TGGTTTGCTACGACGTGGGCTAC-3’ for mouse TNF-α, 5’-GAGGATACCACTCCCAACAGACC-3’ and 5’-AAGTGCATCATCGTTGTTCATACA-3’ for mouse IL-6, 5’-TGTGTCCGTCGTGGATCTGA-3’ and 5’-CCTGCTTCACCACCTTCTTGAT-3’ for mouse GAPDH. Each gene expression level was normalized to that of GAPDH.
Immunofluorescence staining
THP-1 cells were fixed and permeabilized with ice-cold methanol, blocked with 3% BSA-PBS, and incubated with primary antibodies (anti-ASC, anti-cathepsin B, anti-LAMP2, and anti-galectin 3) overnight at 4°C, followed by incubation with secondary antibodies (ASC and LAMP2: goat anti-mouse Alexa Fluor 555, cathepsin B: goat anti-rabbit Alexa Fluor488, and galectin-3: goat anti-rat Alexa Fluor 488, (Invitrogen, San Diego, CA, USA) for 1 hr at room temperature (Noguchi et al., 2023). The immunostained samples were enclosed with Fluoro-KEEPER Antifade Reagent, Non-Hardening Type with DAPI (Nacalai Tesque), and observed using a BZ-X800L fluorescence microscope. For every sample, three images were acquired randomly, and the number, size, and circularity of galectin-3 puncta were measured by ImageJ.
Statistical analysis
The value was expressed as the mean ± standard deviation (S.D.) using Prism software (GraphPad, Boston, MA, USA). All experiments were repeated at least three independent times. Two groups were compared using student’s t-test. Multiple-group comparisons were conducted using the one-way ANOVA analysis of variance followed by the Tukey-Kramer test using Prism software (GraphPad). Data were considered significant when *p < 0.05, **p < 0.01, ***p < 0.001.
RESULTS
BAC promotes mature-IL-1β release in macrophages
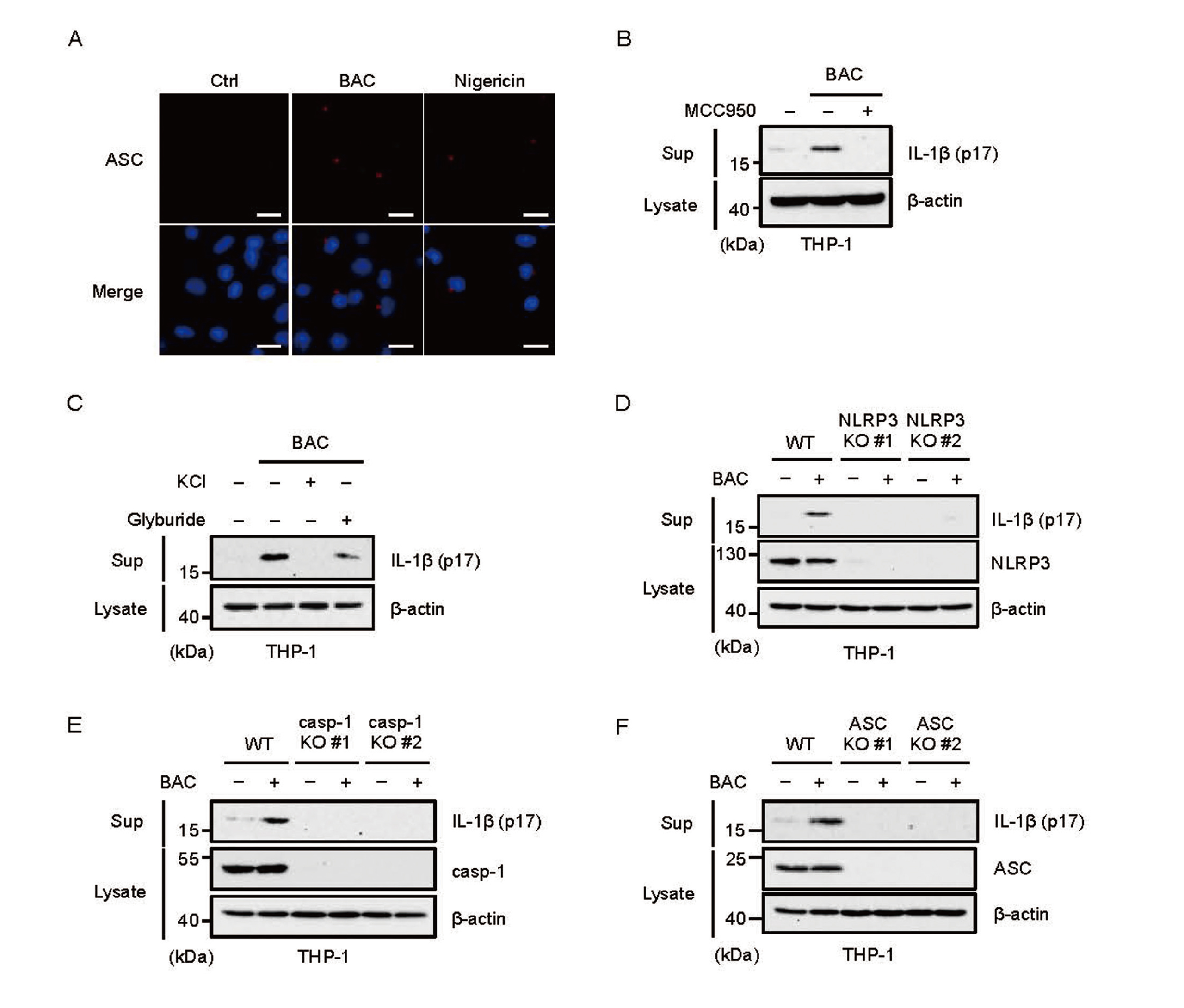
To elucidate the molecular basis for how surfactants initiate inflammation, we focused on the inflammatory responses in macrophages. As shown in Fig. 1A, an enzyme-linked immunosorbent assay (ELISA) revealed that BAC promotes the production of IL-1β, when BAC was used to treat mouse bone marrow-derived macrophages (BMDMs) primed with lipopolysaccharides (LPS), a ligand of toll-like receptor 4 (TLR4) that transcriptionally upregulates inflammatory cytokines. BAC also stimulated the release of IL-1β in human monocytic THP-1 cells that were differentiated into macrophages by phorbol myristate acetate (PMA) (Fig. 1B). Moreover, BAC-induced IL-1β release was confirmed by immunoblot analysis of cell culture supernatants (Fig. 1C). We next tested whether BAC affects pro-IL-1β, the precursor form of IL-1β, expression at mRNA levels, because transcriptional upregulation of pro-IL-1β is an essential step for IL-1β release (Broz and Dixit, 2016). However, BAC failed to upregulate expression of pro-IL-1β, whereas LPS could strongly do so, suggesting that BAC stimulates IL-1β release by activating post-transcriptional steps (Fig. 1D). Moreover, BAC did not upregulate TNF-α and IL-6 production at both protein and mRNA levels (Fig. 1E, 1F, 1G, and 1H). These observations raise the possibility that BAC has the ability to initiate activation of the inflammasomes that govern the post-transcriptional steps of IL-1β release.

We therefore investigated whether BAC activates the inflammasomes. It is known that formation of the ASC speck is a hallmark of inflammasome activation, and its detection by immunofluorescent staining has been well established (Stutz et al., 2013). We thus investigated the ASC speck formation and then found that BAC clearly initiates the ASC speck formation, suggesting that BAC promotes IL-1β release through inflammasome activation (Fig. 2A). Interestingly, we found that BAC-induced IL-1β release was strongly suppressed by the typical NLRP3 inflammasome inhibitors, such as MCC950 (Fig. 2B), and KCl and glyburide (also called glibenclamide) (Fig. 2C). Moreover, knockout of the constituents of the NLRP3 inflammasome, including NLRP3, caspase-1, and ASC, clearly suppressed the BAC-induced IL-1β release (Fig. 2D, 2E and 2F). Therefore, BAC appears to promote the IL-1β release through NLRP3 inflammasome activation.
BAC activates the NLRP3 inflammasome through cathepsin B-dependent mechanisms
We next investigated how BAC triggers NLRP3 inflammasome activation. Interestingly, we found that the cysteine protease inhibitors CA-074 Me and E64d inhibited BAC-induced IL-1β release (Fig. 3A and 3B). Similar results were observed when aluminum hydroxide (alum) adjuvant that induces cathepsin B (CTSB)-dependent NLRP3 inflammasome activation by causing lysosomal rupture, suggesting that BAC-induced IL-1β release is also mediated by lysosomal rupture (Fig. 3C and 3D). To investigate this possibility, we performed immunofluorescence experiments. Under steady-state condition, the lysosome-associated membrane protein-2 (LAMP2), a representative lysosomal marker, and CTSB are colocalized, because CTSB is localized in lysosome (Fig. 3E). However, CTSB diffused in the cytoplasm, when BAC or alum was treated. Moreover, both BAC and alum promoted condensation of galectin-3 (gal-3) induced when lysosomal disorders occur (Fig. 3F). On the other hand, our previous study has demonstrated that lysosomal rupture stimulates NLRP3 activation through the stress-activated kinase c-Jun N-terminal kinase (JNK) (Okada et al., 2014). However, BAC failed to activate JNK, whereas LPS strongly activated (Fig. 3G). Taken together, these observations suggest that BAC causes lysosomal rupture that promotes leakage of CTSB to cytoplasm, and thereby activates the NLRP3 inflammasome, similarly to alum.
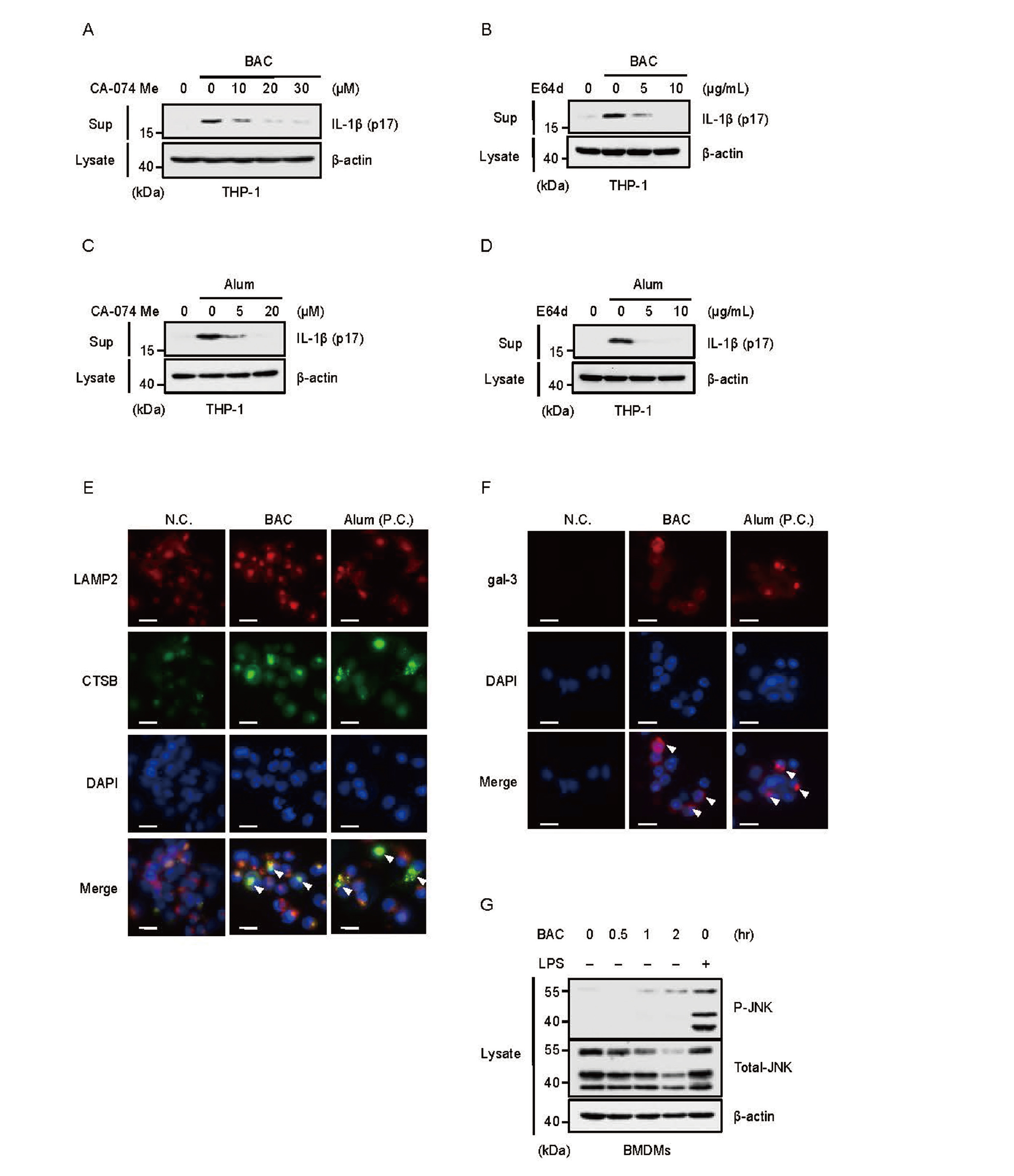
We next investigated how BAC triggers lysosomal rupture. In this regard, we speculated that BAC causes plasma membrane disturbance, and thereby transmembrane ion fluxes. Indeed, BAC increased cytosolic Ca2+ levels when analyzing Ca2+ using the calcium-sensitive dye Fluo 4-AM (Fig. 4A). Interestingly, BAC failed to stimulate the IL-1β release in PMA-primed THP-1 cells cultured in Ca2+ free media (Fig. 4B), and the addition of CaCl2 but not MgCl2 to Ca2+ free media successfully recovered BAC-induced IL-1β release (Fig. 4C). Moreover, the calcium chelator BAPTA-AM suppressed BAC-induced IL-1β release (Fig. 4D). Therefore, we next examined relations between Ca2+ influx and lysosomal rupture responsible for BAC-induced NLRP3 inflammasome activation. As shown in Fig. 4E and 4F, BAPTA-AM strongly inhibited condensation of galectin-3. Taken together, these results suggest that Ca2+ influx induced by BAC initiates lysosomal rupture.
Conventional surfactants can activate the NLRP3 inflammasome
Finally, we tested whether conventional surfactants other than BAC can activate the NLRP3 inflammasome. As we expected, all typical surfactants, such as sodium dodecyl sulfate (SDS), the natural surfactant saponin, and Triton X-100, promoted IL-1β release, which was inhibited in NLRP3 knockout THP-1 cells (Fig. 5A-5C). Of note, the NLRP3 expression was decreased when the surfactants were treated (Fig. 2D and Fig. 5A-C). These observations raise the possibility that the NLRP3 inflammasome activation induced by the surfactants stimulates the negative feedback mechanisms that have been demonstrated previously (Song et al., 2016; Yan et al., 2015). Taken together, these results suggest that these conventional surfactants other than BAC also activate the NLRP3 inflammasome through the same mechanisms as BAC (Fig. 5D).
DISCUSSION
In this study, we aimed to clarify the mechanisms by which BAC causes health hazards such as interstitial pneumonia by focusing inflammatory responses in macrophages. As a result, we found that BAC triggers lysosomal destruction, and release of cathepsin B into the cytoplasm by increasing the cytosolic Ca2+ levels, which thereby induces NLRP3 inflammasome activation and subsequent inflammation. In addition, we found that other surfactants, treated at a concentration that was determined based on a previous report (Mizutani et al., 2016) and our own preliminary studies, also activate the NLRP3 inflammasome (Fig. 5A-C). Although how Ca2+ influx induced by BAC causes lysosomal rupture is not elucidated, Ca2+ influx-mediated lysosomal rupture may be a common mechanism of the NLRP3 inflammasome activation initiated by conventional surfactants.
Our results revealed that BAC directly acts on macrophages, and stimulates the release of mature IL-1β. However, since macrophages are basically present in blood vessels, it is not yet clear how macrophages are exposed to BAC. In this regard, since the surfactants have a membrane-disrupting effect, the surfactants may infiltrate epithelial tissues, reach blood vessels, and then macrophages are exposed to the surfactants (Parsi, 2015). Thereafter, mature IL-1β is released from macrophages, which initiates inflammation associated with the BAC-related health hazards. On the other hand, BAC is widely used in eye drops as a preservative, and is known to have toxic and pro-inflammatory effects on ocular surface tissues (Michée et al., 2013; Baudouin et al., 2010; Nuzzi et al., 1995). Exposure to BAC at a concentration of 329 nM (10-4%) has been reported to inhibit proliferation and induce apoptosis in conjunctival and corneal epithelial cells (Ammar et al., 2011; Brasnu et al., 2008). Therefore, even when exposed to low concentrations of BAC, tissue defects due to the inhibition of epithelial cell proliferation and induction of apoptosis may facilitate the exposure of BAC to macrophages, which initiates the release of the proinflammatory cytokine IL-1β. Thus, although further studies are necessary to certify the mechanisms by which conventional surfactants initiate inflammatory diseases, our findings reveal a novel pro-inflammatory action of the surfactants, which may lead to a better understanding of inflammatory lesions, such as HDLI, induced by the surfactants.
ACKNOWLEDGMENTS
We thank all members of Lab of Health Chemistry for helpful discussions. This work was supported by JSPS KAKENHI Grant Numbers JP21H02691, JP21H02620, JP22J20257, JP24K02237 and JP24K02173. This work was also supported by the Division for Interdisciplinary Advanced Research and Education (DIARE) Tohoku University.
Conflict of interest
The authors declare that there is no conflict of interest.
REFERENCES
- Ammar, D.A., Noecker, R.J. and Kahook, M.Y. (2011): Effects of benzalkonium chloride- and polyquad-preserved combination glaucoma medications on cultured human ocular surface cells. Adv. Ther., 28, 501-510.
- Basketter, D.A., Marriott, M., Gilmour, N.J. and White, I.R. (2004): Strong irritants masquerading as skin allergens: the case of benzalkonium chloride. Contact Dermatitis, 50, 213-217.
- Baudouin, C., Labbé, A., Liang, H., Pauly, A. and Brignole-Baudouin, F. (2010): Preservatives in eyedrops: the good, the bad and the ugly. Prog. Retin. Eye Res., 29, 312-334.
- Brasnu, E., Brignole-Baudouin, F., Riancho, L., Warnet, J.M. and Baudouin, C. (2008): Comparative study on the cytotoxic effects of benzalkonium chloride on the Wong-Kilbourne derivative of Chang conjunctival and IOBA-NHC cell lines. Mol. Vis., 14, 394-402.
- Broz, P. and Dixit, V.M. (2016): Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol., 16, 407-420.
- Choi, H.Y., Lee, Y.H., Lim, C.H., Kim, Y.S., Lee, I.S., Jo, J.M., Lee, H.Y., Cha, H.G., Woo, H.J. and Seo, D.S. (2020): Assessment of respiratory and systemic toxicity of Benzalkonium chloride following a 14-day inhalation study in rats. Part. Fibre Toxicol., 17, 5.
- Jeon, H., Kim, D., Yoo, J. and Kwon, S. (2019): Effects of benzalkonium chloride on cell viability, inflammatory response, and oxidative stress of human alveolar epithelial cells cultured in a dynamic culture condition. Toxicol. In Vitro, 59, 221-227.
- Kagi, T., Inoue, A., Noguchi, T., Suzuki, W., Takano, S., Otani, K., Naganuma, R., Sekiguchi, Y., Hirata, Y., Shindo, S., Hwang, G.W. and Matsuzawa, A. (2024): The NLRP3 Inflammasome Is a Major Cause of Acute Renal Failure Induced by Polypeptide Antibiotics. J. Immunol., 212, 1807-1818.
- Kagi, T., Naganuma, R., Inoue, A., Noguchi, T., Hamano, S., Sekiguchi, Y., Hwang, G.W., Hirata, Y. and Matsuzawa, A. (2022): The polypeptide antibiotic polymyxin B acts as a pro-inflammatory irritant by preferentially targeting macrophages. J. Antibiot. (Tokyo), 75, 29-39.
- Kagi, T., Noguchi, T. and Matsuzawa, A. (2021): Mechanisms of gefitinib-induced interstitial pneumonitis: why and how the TKI perturbs innate immune systems? Oncotarget, 12, 1321-1322.
- Kelley, N., Jeltema, D., Duan, Y. and He, Y. (2019): The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci., 20.
- Lavorgna, M., Russo, C., D’Abrosca, B., Parrella, A. and Isidori, M. (2016): Toxicity and genotoxicity of the quaternary ammonium compound benzalkonium chloride (BAC) using and as model systems. Environ. Pollut., 210, 34-39.
- Ling, T.C. and Highet, A.S. (2000): Irritant reactions to an antiseptic bath emollient. J. Dermatolog. Treat., 11, 263-267.
- Michée, S., Brignole-Baudouin, F., Riancho, L., Rostene, W., Baudouin, C. and Labbé, A. (2013): Effects of benzalkonium chloride on THP-1 differentiated macrophages in vitro. PLoS One, 8, e72459.
- Mizutani, T., Mori, R., Hirayama, M., Sagawa, Y., Shimizu, K., Okano, Y. and Masaki, H. (2016): Sodium Lauryl Sulfate Stimulates the Generation of Reactive Oxygen Species through Interactions with Cell Membranes. J. Oleo Sci., 65, 993-1001.
- Noguchi, T., Sekiguchi, Y., Kudoh, Y., Naganuma, R., Kagi, T., Nishidate, A., Maeda, K., Ishii, C., Toyama, T., Hirata, Y., Hwang, G.W. and Matsuzawa, A. (2021): Gefitinib initiates sterile inflammation by promoting IL-1β and HMGB1 release via two distinct mechanisms. Cell Death Dis., 12, 49.
- Noguchi, T., Sekiguchi, Y., Shimada, T., Suzuki, W., Yokosawa, T., Itoh, T., Yamada, M., Suzuki, M., Kurokawa, R., Hirata, Y. and Matsuzawa, A. (2023): LLPS of SQSTM1/p62 and NBR1 as outcomes of lysosomal stress response limits cancer cell metastasis. Proc. Natl. Acad. Sci. USA, 120, e2311282120.
- Noguchi, T., Suzuki, M., Mutoh, N., Hirata, Y., Tsuchida, M., Miyagawa, S., Hwang, G.W., Aoki, J. and Matsuzawa, A. (2018): Nuclear-accumulated SQSTM1/p62-based ALIS act as microdomains sensing cellular stresses and triggering oxidative stress-induced parthanatos. Cell Death Dis., 9, 1193.
- Nuzzi, R., Vercelli, A., Finazzo, C. and Cracco, C. (1995): Conjunctiva and subconjunctival tissue in primary open-angle glaucoma after long-term topical treatment: an immunohistochemical and ultrastructural study. Graefes Arch. Clin. Exp. Ophthalmol., 233, 154-162.
- Okada, M., Matsuzawa, A., Yoshimura, A. and Ichijo, H. (2014): The lysosome rupture-activated TAK1-JNK pathway regulates NLRP3 inflammasome activation. J. Biol. Chem., 289, 32926-32936.
- Parsi, K. (2015): Interaction of detergent sclerosants with cell membranes. Phlebology, 30, 306-315.
- Presley, C.L., Militello, M., Barber, C., Ladd, R., Laughter, M., Ferguson, H., Dewey, J., Pulsipher, K.J., Rundle, C.W. and Dunnick, C.A. (2021): The History of Surfactants and Review of Their Allergic and Irritant Properties. Dermatitis, 32, 289-297.
- Sekiguchi, Y., Takano, S., Noguchi, T., et al. (2023): The NLRP3 Inflammasome Works as a Sensor for Detecting Hypoactivity of the Mitochondrial Src Family Kinases. The Journal of Immunology.
- Sekiguchi, Y., Yamada, M., Noguchi, T., Noomote, C., Tsuchida, M., Kudoh, Y., Hirata, Y. and Matsuzawa, A. (2019): The anti-cancer drug gefitinib accelerates Fas-mediated apoptosis by enhancing caspase-8 activation in cancer cells. J. Toxicol. Sci., 44, 435-440.
- Song, H., Liu, B., Huai, W., Yu, Z., Wang, W., Zhao, J., Han, L., Jiang, G., Zhang, L., Gao, C. and Zhao, W. (2016): The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat. Commun., 7, 13727.
- Song, J.H., Ahn, J., Park, M.Y., Park, J., Lee, Y.M., Myong, J.P., Koo, J.W. and Lee, J. (2022): Health Effects Associated With Humidifier Disinfectant Use: A Systematic Review for Exploration. J. Korean Med. Sci., 37, e257.
- Stutz, A., Horvath, G.L., Monks, B.G. and Latz, E. (2013): ASC speck formation as a readout for inflammasome activation. Methods Mol. Biol., 1040, 91-101.
- Tsuchida, M., Yokosawa, T., Noguchi, T., Shimada, T., Yamada, M., Sekiguchi, Y., Hirata, Y. and Matsuzawa, A. (2020): Pro-apoptotic functions of TRAF2 in p53-mediated apoptosis induced by cisplatin. J. Toxicol. Sci., 45, 219-226.
- Yan, Y., Jiang, W., Liu, L., Wang, X., Ding, C., Tian, Z. and Zhou, R. (2015): Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell, 160, 62-73.