Abstract
Hypoxia induces the expression of nuclear factor kappa B (NF-kappa-B). NF-kappa-B functions by forming dimers from five main subunits: p65 (RelA), RelB, p52, p50, and c-Rel. In the classical pathway, NF-kappa-B activity is regulated by the degradation-inducing factor I kappa B kinase (IKK). IKK is composed of an α/β isomer and essential modulator NEMO (γ) subunits in the classical pathway, which may be the major pathway for NF-kappa-B signaling. In the present study, we focused on factor-inhibiting HIF-1 (FIH-1) and Prolyl hydroxylase domain enzyme (PHD), which have been identified as oxygen concentration-dependent regulators of HIF-1α. PHD has three isoforms: PHD1, PHD2, and PHD3, which have different affinities towards HIF-1α. We examined the interactions between IKKα/β and PHD1-3 by immunoprecipitation. PHDs efficiently interacted with IKKα/β. Furthermore, the overexpression of PHDs decreased the mRNA level of IL-1β, a downstream factor of NF-kappa-B activated by LPS. The overexpression of PHD1 and PHD2 markedly reduced IKKα/β protein levels; however, the effects of PHD3 were weaker than those of PHD1 and PHD2. Mutants of the active sites of PHD1 and PHD2 did not decrease IKKα/β protein levels, and a mutation in the active site of PHD3 did not affect IKKα/β protein levels. We also attempted to investigate the interactions of FIH-1 with IKKα/β and IκBα by immunoprecipitation, but found none. Moreover, IKKα/β and p65 protein levels were not affected by the overexpression of FIH-1. Collectively, these results suggest that PHDs directly regulated IKK protein levels, while FIH-1 did not affect the NF-kappa-B classical pathway.
INTRODUCTION
Hypoxia is closely associated with various pathological conditions, such as cancer and ischemic diseases (Prabhakar and Semenza, 2015). The key factor acting in hypoxia is hypoxia-inducible factor (HIF)-1α (Laughner et al., 2001; Fukuda et al., 2002). The expression and activity of HIF-1α are regulated by the oxygen concentration-dependent enzymes prolyl hydroxylase domain protein (PHD) and factor-inhibiting HIF-1 (FIH-1). Under normoxic conditions, HIF-1α is hydroxylated at the 402nd and 564th proline residues by PHD with oxygen as a cofactor. The resulting hydroxylation of HIF-1α is a key step in the degradation of HIF-1α. As a result, the conformation of HIF-1α changes and HIF-1α binds to von Hippel-Lindau tumor suppressor protein (pVHL). pVHL is part of E3 ubiquitin ligase (Maxwell et al., 2001). In addition, asparagine residues near the C terminus are hydroxylated by FIH-1, which uses oxygen as a cofactor. The hydroxylation of HIF-1α at the 803rd Asn residue inhibits the interaction of HIF-1α with CBP/p300, resulting in transcriptional repression. Under normoxic conditions, the transcriptional activity of HIF-1α is largely suppressed despite HIF-1α being constantly biosynthesized, which maintains the intracellular expression and transcriptional activity of HIF-1α at a low level (Maxwell et al., 1999; Ohh et al., 2000; Lee et al., 2004). Stabilized HIF-1α under hypoxic conditions translocates into the nucleus, forms a dimer with the aryl hydrocarbon nuclear translocator, and then binds to the hypoxia-response element in order to induce the transcription of hypoxia-responsive genes, such as vascular endothelial growth factor (VEGF) and erythropoiesis-regulating hormone (EPO) (Semenza, 2001; Dames et al., 2002; Freedman et al., 2002; Schofield and Ratcliffe, 2004).
Signaling pathways from receptors to nuclear factor-kappa B (NF-kappa-B) are involved in inflammatory responses (Karin, 1999; Tergaonkar, 2006; Gilmore, 2006; Scheidereit, 2006). NF-kappa-B, which functions by forming dimers from five main subunits: p65 (RelA), RelB, p52, p50, and c-Rel, plays a central role in the various changes that occur in inflammation, such as cytokine and chemokine transactivation followed by the promotion of macrophage filtration (Gilmore, 2006). Various stimuli, including lipopolysaccharide (LPS), have been shown to activate NF-kappa-B (Perkins, 2012). In a representative pathway, such as the conventional NF-kappa-B pathway, NF-kappa-B is composed of the p50 and p65 (RelA) subunits and the nuclear translocation of NF-kappa-B occurs through the activation of inhibitor of NF-kappa-B (IkB) kinase (IKK) followed by the proteasomal degradation of IkB. IKK, a serine kinase, is composed of three subunits: IKKα, IKKβ, and IKKγ, with IKKα and IKKβ functioning as kinase subunits and IKKγ being a regulatory subunit (NEMO) of the IKK complex (DiDonato et al., 1997). IKK phosphorylates IkB, followed by the proteasomal degradation of IkB, which promotes the nuclear translocation of NF-kappa-B, thereby activating inflammatory responses through the transactivation of inflammatory cytokines, such as interleukin (IL)-1β (Hayden and Ghosh, 2008; Hinz and Scheidereit, 2014).
The regulatory mechanism of NF-kappa-B activity is complex, and previous studies reported cross-talk between the immune response and hypoxic response systems (Rius et al., 2008). At the site of inflammation, tissue becomes hypoxic not only because filtrated immune cells consume oxygen, but also because impaired vascular blood flow impedes the provision of oxygen. The activity of NF-kappa-B was also shown to be affected by PHD through its inhibitory effects on the interaction between IKKβ and Heat shock protein 90 (Hsp90), which suppressed the activation of IKKβ (Xue et al., 2010). Furthermore, PHD hydroxylated IKK (Strowitzki et al., 2019), while FIH hydroxylated IkB, but did not affect NF-kappa-B activity (Cockman et al., 2006). However, the mechanisms responsible for the effects of FIH-1 and each isoform of PHD on the NF-kappa-B pathway remain unclear. In the present study, we investigated the substrate specificity and active sites of PHD1, 2, and 3 for IKKα/β based on comparisons with HIF-1α and the activity of FIH towards IKKα/β
MATERIALS AND METHODS
Cell culture and treatment of cells with reagents
Human cervical carcinoma HeLa cells were provided by Prof. S.T. Suzuki (Kwansei Gakuin University). They were grown in high-glucose Dulbecco’s Modified Eagle’s Medium (Wako Pure Chemicals, Osaka, Japan) containing 10% fetal bovine serum (Sigma-Aldrich) and 1% penicillin-streptomycin (Sigma-Aldrich) and cultured at 37°C, 5% CO2. HeLa cells were treated with 5 µM MG132 (Wako), a proteasomal inhibitor, and 200 ng/mL LPS or with 100 µM dipyridyl (Wako), an inhibitor of PHD, for 6 hr.
RNA extraction and RT-PCR
HeLa cells were cultured at 2.0 × 105 cells in 6-cm Petri dishes and washed once with ice-cold PBS. Total RNA was isolated with Isogen II (Takara) and converted to cDNA with a reverse transcriptase kit (Takara) according the manufacturer’s instructions. cDNAs were used for PCR.
Preparation of constructs for overexpression
Human FIH-1 cDNA (Acc No. NM_017902.3) was isolated by PCR using cDNA as a template with KOD Fx neo (Toyobo, Tokyo, Japan) with the primers (Table 1) and conditions shown below. The resulting PCR product was digested with BamHI and EcoRI and inserted into the BamHI and EcoRI sites of the FLAG-pcDNA4 vector (Siswanto et al., 2023). PCR conditions were as follows: at 94°C for 2 min and 30 cycles at 94°C for 30 sec, 55°C for 30 sec, and then 68°C for 1 min 10 sec. The full-length cDNAs of human PHD1 (NM_053046.3), PHD2 (NM_022051.2), and PHD3 (NM_001308103.1) were amplified by PCR (KOD Fx neo) using cDNA as the template with primers (Table 1). The resulting PCR product was digested with EcoRI and XhoI and inserted into the EcoRI and XhoI sites of the FLAG-pcDNA4 vector (Siswanto et al., 2023). PCR conditions were as follows: at 94°C for 2 min and 30 cycles at 94°C for 30 sec, 55°C for 30 sec, and then 68°C for 1 min.
Table 1. Primers for the construction of full-length FIH and PHD1-3 cDNAs.

Preparation of active site mutants of PHD1-3 constructs for overexpression
PHD1/FLAGpcDNA4, PHD2/FLAGpcDNA4, and PHD3/FLAGpcDNA4 prepared above were used as templates. Primers (Table 2) were used for PCR to replace arginine with alanine in the 2-oxoglutaric acid binding site, one of the cofactors of each PHD (Tarhonskaya et al., 2014). The former and latter halves of the PHD1 mutant were prepared with forward primer 1 and reverse primer 2 and with forward primer 3 and reverse primer 4, respectively. The former and latter halves of the PHD1 mutant were prepared with forward primer 5 and reverse primer 6 and with forward primer 7 and reverse primer 8, respectively. The former and latter halves of the PHD3 mutant were prepared with forward primer 9 and reverse primer 10 and with forward primer 11 and reverse primer 12, respectively. Former and latter fragments were each mixed and the full-length DNAs of PHDs were amplified by PCR. PCR was performed at 94°C for 2 min and 30 cycles at 94°C for 30 sec, 58°C for 30 sec, and 68°C for 1 min. Full-length cDNAs were subjected to a restriction enzyme treatment, ligated using DNA Ligation Kit ver.1 (Takara Bio), and transformed using DH5α competent cells (Toyobo)
Table 2. Primers for the preparation of constructs for active site mutants of PHD1-3.

Immunoprecipitation method
Two 10-cm dishes of FIH-1- or PHD-overexpressing cells by the calcium-phosphate method were washed with 5 mL of cooled PBS, 300 µL of IP buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% NP40, 5 mM EDTA, and 1% PI cocktail) was added, and cells were incubated on ice for 5 min. Cells were then collected in a 1.5-mL tube with a cell scraper and centrifuged at 12,000 rpm at 4°C for 15 min. The supernatant was decanted into a new 1.5-mL tube. After quantifying the amount of protein by the BCA method, 500 µg of protein was taken into a new 1.5-mL tube and made up to 500 µg/500 µL with IP buffer. One microgram of an anti-DYKDDDDK (Flag) tag antibody (Wako Pure Chemicals, Osaka, Japan: 1000-fold dilution) or anti-mouse control IgG (prepared in our laboratory) was added, and the tube was stirred at 4°C overnight. Twenty microliters of Protein G Sepharose 4 Fast Flow (Cytiva, Tokyo) was added and the tube was stirred at 4°C for 1 hr. After centrifugation at 5,000 rpm at 4°C for 3 min, the supernatant was decanted into a new 1.5-mL tube and this was used as the flowthrough sample. One milliliter of wash buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, and 0.05% NP-40) was added to wash the beads. The tube was centrifuged at 5000 rpm at 4°C for 3 min and the supernatant was discarded. This process to wash was repeated three times. In the first elution, 16.7 µL of 0.1 M glycine buffer pH 2.5 was added and incubated at 37°C for 15 min for acid elution; following centrifugation at 5000 rpm, the supernatant was transferred to a new 1.5-mL tube, and 3.3 µL of 6×sample buffer and 1 M Tris-HCL pH 9.0 were added until the sample buffer turned blue-purple. The sample was incubated at 95°C for 5 min for SDS-PAGE. Twenty microliters of 1×sample buffer was added to the 1.5-mL tube with Protein G Sepharose 4 Fast Flow, which was directly treated in the same manner as the first elution. The sample was incubated at 95°C for 5 min for denaturation.
Semi-quantitative PCR
The primer sequences for IL-1β (NM_000576.3) and β-actin (NM_000963.4) are shown in Table 3. PCR conditions were as follows: at 95°C for 2 min and 30 cycles at 95°C for 30 sec, 55°C for 30 min, and 72°C for 30 min. The PCR products obtained were electrophoresed on a 1% agarose gel (NIPPON GENE, Tokyo) containing TAE buffer (40 mM Tris, 20 mM acetic acid, and 1 mM EDTA) and bands were visualized using AE-6905H Image Save HR (ATTO). Images were quantified using Image J.
Table 3. Primers for RT-PCR.

SDS-PAGE and Western Blotting
Samples were electrophoresed using acrylamide gels. After electrophoresis, gels were immersed in transfer buffer (100 mM Tris, pH 7.5, 190 mM glycine, and 20% methanol) and shaken for 15 min. The gels were transferred to a nitrocellulose membrane by semi-dry blotting. Membranes were blocked in 2% skim milk in TPBS (PBS with 0.05% Tween-20) for 30 min by shaking. Membranes and primary antibodies were reacted at 4°C overnight. Membranes were washed three times with TPBS for 10 min. Membranes and secondary antibodies were reacted at room temperature for 45 min. After the second antibody reaction, membranes were washed three times with PBS for 10 min and reacted with 4-chloro-1-naphtathol to visualize bands. Regarding the primary antibodies, an anti-actin antibody (produced in our laboratory, 3000× dilution) was diluted in antibody diluent (0.1% BSA in TPBS). An anti-DYKDDDDK tag antibody (Wako Pure Chemicals, Osaka, Japan) and anti-IKK antibody (Abcam, UK), which recognizes both IKKα and IKKβ, was diluted 1000 times with Immuno-Enhancer Reagent A (Wako Pure Chemicals, Osaka, Japan). The anti-RelA (p65) antibody, which was prepared in our laboratory (Siswanto et al., 2023), was diluted 1000 times with Immuno-Enhancer Reagent A. The second antibody, goat IgG (H+L)-HRP was diluted with the same diluent as the primary antibody (Antibody Diluent or Immuno-Enhancer Reagent B). When we expected higher sensitivity, we used the chemiluminescence method. Regarding detection by chemiluminescence, the PVDF membrane blotted with proteins was immersed in Immobilon Western Chemiluminescent HRP Substrate (Sigma-Aldrich) for 5 min and detection was performed with LAS-X (Fuji Chemical Company, Tokyo, Japan).
Statistical analysis
Statistical analyses for single comparisons between means were performed using the Student’s t-test. An analysis of variance for multiple comparisons and the Bonferroni-Dunn test as a post-hoc test were performed using StatView.
RESULTS
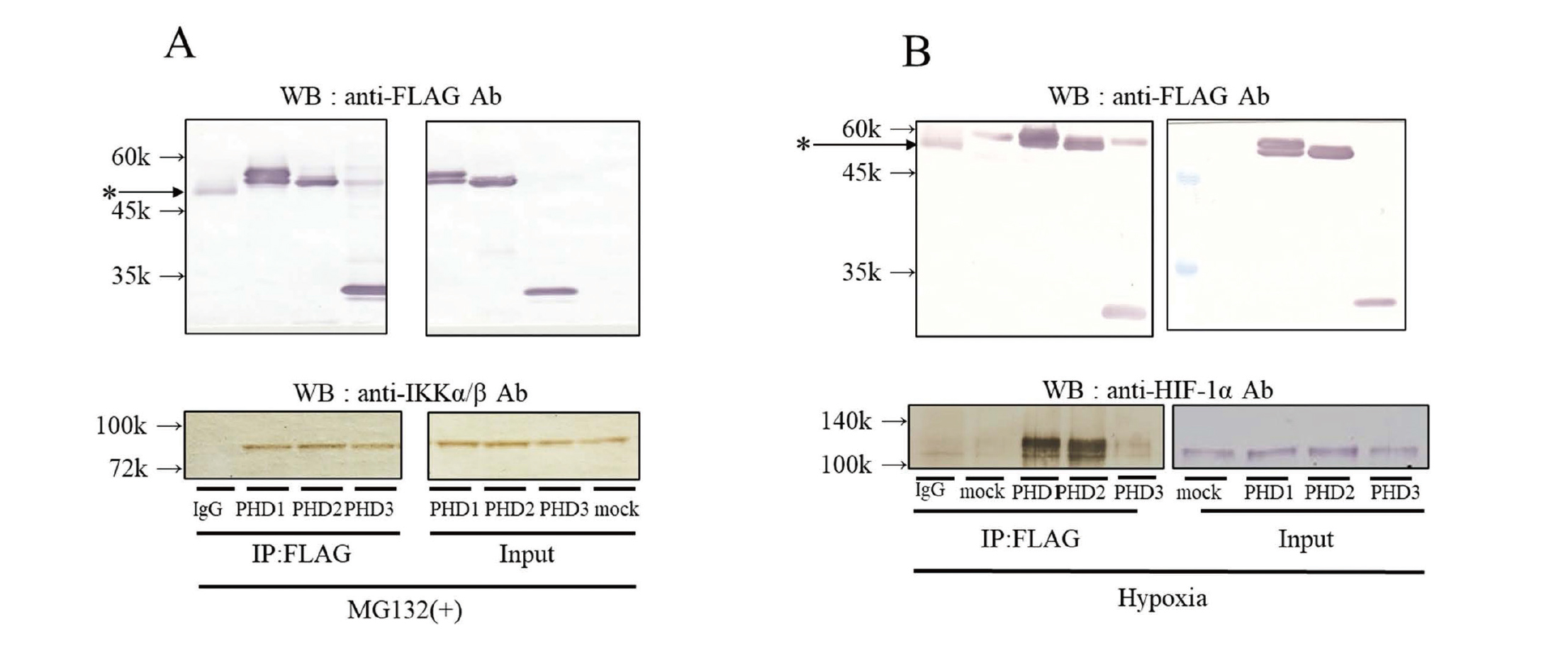
Interaction of Flag-fused PHD1-3 in HeLa cells with IKKα/β and HIF-1α
PHD3 has been reported to interact with IKK (Xue et al., 2010); however, the affinity and interaction of each PHD isoform with IKK remain unclear. Therefore, we overexpressed Flag-fused PHD1-3 in HeLa cells and investigated whether each PHD isoform interacted with IKKα/β and HIF-1α as a positive control (Fig. 1). To avoid the proteasomal degradation of HIF-1α and IKK, we used MG132 during immunoprecipitation. In the presence of MG132, the sedimentation of PHD1-3 showed almost the same amounts of the IKKα/β proteins. On the other hand, under hypoxic conditions, the sedimentation of PHD1 and PHD2 resulted in the same amount of HIF-1α, while PHD3 sedimented less HIF-1α even though its own sedimentation amount was the same. PHD3 interacted less with HIF-1α than PHD1 and 2.
Changes in levels of IKKα/β proteins and IL-1β mRNA, a downstream factor of NF-kappa-B, upon the overexpression of PHD1-3 in HeLa cells
We confirmed that hypoxia (a low oxygen concentration) enhanced the expression of LPS-induced IL-1β mRNA in HeLa cells (Supplemental Fig. 1) and also induced IL-1β mRNA as well as CA9 mRNA regulated by HIF-1α. Flag-PHD1-3 were then overexpressed in HeLa cells. The IKKα/β protein levels tended to decrease but it is not significant (Supplemental Fig. 2A). Treatment of HeLa cells with LPS did not change IKKα/β protein levels compared with control (Supplemental Fig. 2B). Next, changes in the levels of the IKKα/β proteins and IL-1β mRNA, a downstream factor of NF-kappa-B, were examined after the treatment of cells with LPS (Fig. 2). IKKα/β protein levels were markedly lower after the overexpression of PHD1-3 than in controls, and PHD1 and 2 exerted stronger effects than PHD3. IL-1β mRNA up-regulated by LPS was more effectively reduced by the overexpression of PHD1 and 2 than by that of PHD3; however, no significant difference was noted in the strength of PHD1, 2, or 3 interactions with IKKα/β.

Since PHD1 and 2 or PHD3 exerted different effects on IKK and the NF-kappa-B signaling pathway when PHDs were overexpressed, we overexpressed mutant PHD1-3, not exhibiting enzymatic activity, to compare their effects with that of the wild type (Fig. 3). Decreases in the protein levels of IKKα/β were abolished by the PHD1 and PHD2 mutants, but not by the PHD3 mutant. This result suggests that PHD1 and 2 mainly acted on IKKα/β, while the effects of PHD3 were weak and, thus, their activity is important in the regulation of IKK.
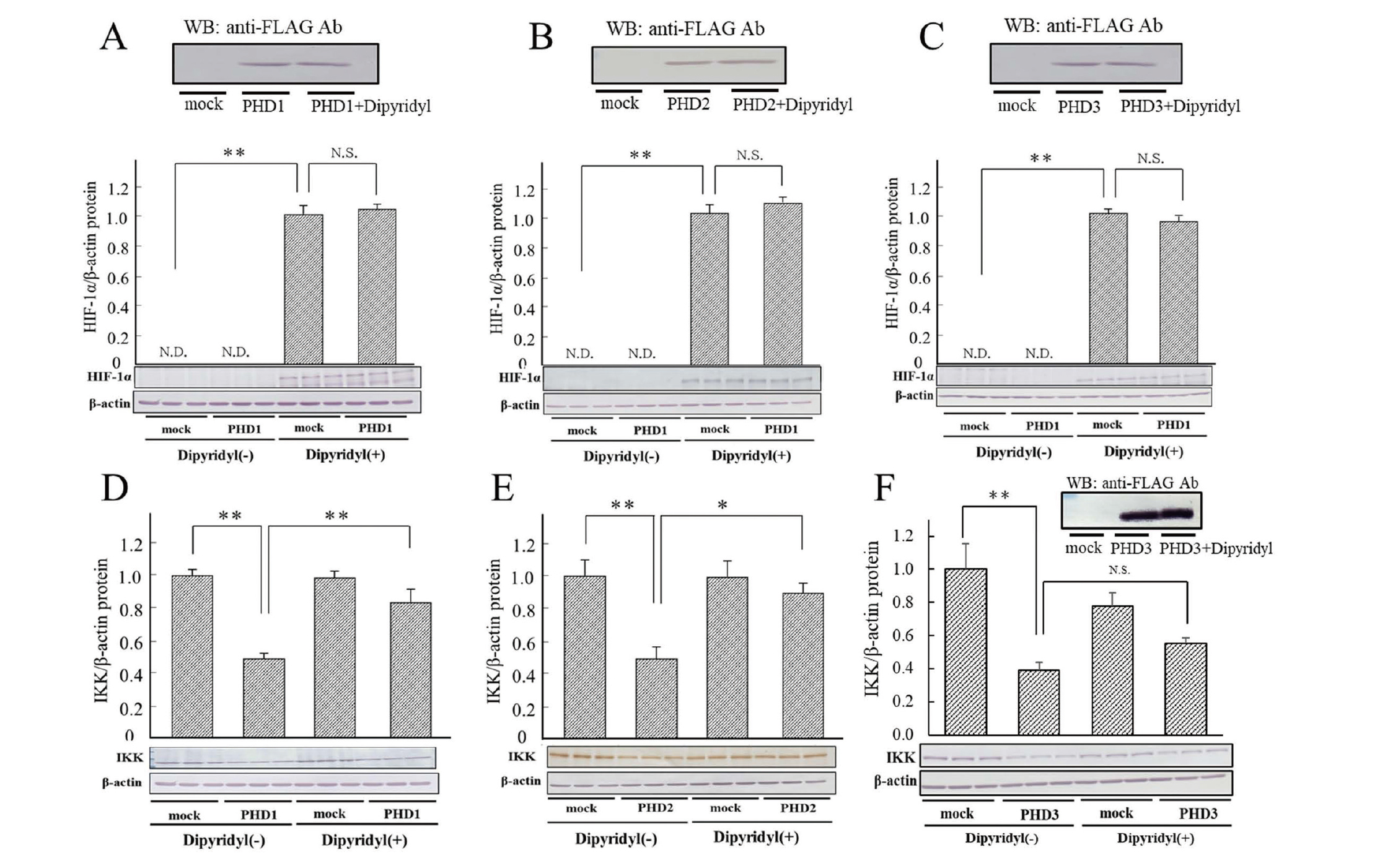
Recovery of decreases in IKKα/β protein levels by the overexpression of PHD1-3 with dipyridyl and MG132
HIF-1α is hydroxylated by PHD1-3 and degraded by the ubiquitin-proteasome system. PHDs require Fe2+ and 2-oxoglutarate to oxidize a substrate. In the present study, the effects of dipyridyl (Fig. 4), an iron chelator and inhibitor of PHDs, and MG132 (Fig. 5), a proteasome inhibitor, on IKKα/β and HIF-1α protein levels as a positive control under the overexpression of PHD1-3 were investigated. The addition of dipyridyl significantly recovered decreases in IKKα/β protein levels caused by the overexpression of PHD1 and PHD2, but not PHD3. Taken together with the results obtained from active site mutants, PHD activity was important in the regulation of IKK. Furthermore, the addition of MG132 markedly recovered the decreases induced in IKKα/β levels by the overexpression of PHD1 and PHD2, but not PHD3. These results suggest that PHD1 and 2 hydroxylated IKK, oxidized IKK was degraded by proteasomes, and PHD1 and PHD2 exerted different effects on IKK from PHD3; however, the confirmation of the direct oxidation of IKK by PHD may be necessary.

FIH belongs to the same family as PHD and plays an important role in the regulation of HIF-1α activity. On the other hand, FIH has been shown to oxidize IκBα (Cockman et al., 2006; Strowitzki et al., 2019). Therefore, we overexpressed Flag-fused FIH-1 in HeLa cells and examined its interaction with IKKα/β and IκBα by immunoprecipitation (Fig. 6). The interaction of FIH-1 with IkBα was previously reported (Cockman et al., 2006). Detection by chemiluminescence, which has higher sensitivity than staining with 4-chloro-1-naphthol, was performed but interaction was not detected.
The results obtained showed that neither IKKα nor IKKβ was precipitated, suggesting no interaction with FIH-1. Furthermore, the overexpression of FIH-1 did not affect IKKα/β or p65 protein expression levels (Fig. 7). These results indicate that FIH-1, unlike PHD1-3, did not act on the IKK-IkB pathway.
DISCUSSION
Hypoxia is closely associated with inflammation and immune responses (Prabhakar and Semenza, 2015). In the hypoxic response system, PHDs hydroxylate HIF-1α in an oxygen-dependent manner and suppress the transcription of its downstream factor, EPO, suggesting that IKK functions may be suppressed by PHDs in the innate immune response system (D'Ignazio and Rocha, 2016). Since PHDs require oxygen to function, we examined the effects of PHDs on the innate immune response system and found that PHD1-3 differentially regulated IKK, a key factor in the NF-kappa-B pathway. We investigated whether PHDs interacted directly with HIF-1α and IKK, and found interactions for all isoforms, but to different degrees. Hypoxia and inflammatory responses are closely related, and since NF-kappa-B induces the expression of HIF-1α, which, in turn, induces that of NF-kappa-B(p65) and IKK, hypoxic and inflammatory responses have been suggested to trigger a positive feedback loop (Rius et al., 2008).
The hydroxylation of the proline residues of HIF has been identified as the major function of PHD; however, PHD may have regulatory functions other than hydroxylation (Maxwell et al., 1999). To investigate whether the regulation of IKK by each PHD isoform was due to the hydroxylation activities of PHDs, we examined the expression levels of HIF-1α and IKK following the overexpression of PHDs with mutations in the 2-oxoglutarate-binding site and a treatment with a PHD inhibitor. HIF-1α expression levels were effectively decreased by the overexpression of PHD1 and PHD2, but not PHD3 under hypoxic conditions (Supplemental Fig. 3). IKKα/β protein levels also decreased when the PHD1 and PHD2 isoforms were overexpressed, but not when mutant PHDs were overexpressed. Furthermore, dipyridyl, a PHD inhibitor, recovered the decreased expression of IKKα/β . IKK protein expression levels when PHD3 was overexpressed were low and the contribution of PHD3 activity to the regulation of IKKα/β protein expression was small, suggesting that the activity of PHD3 was not necessary to inhibit IKK protein expression. As reported by other researchers (Nakayama et al., 2007; Strowitzki et al., 2019), the PHD1-PHD3 or PHD2-PHD3 heterodimer is formed. Considering these results, we can assume that PHD3 supports the activity of PHD1 and PHD2, and that the active site of PHD3 is not involved.
We examined IKK expression levels when PHD isoforms were overexpressed together with the addition of MG132, a proteasome inhibitor. The results obtained showed that MG132 restored IKK expression levels, suggesting that the degradation of IKK by PHD occurred in a proteasome-dependent manner. These results indicate that PHD1 and PHD2 exerted similar effects on HIF-1α and IKK. The N-terminal regions of PHD1 and 2, which are absent in PHD3, may be important for their effects on hypoxic and innate immune responses (Nakayama et al., 2007).
There is evidence for complex crosstalk or feedback between NF-kappa-B and HIF-1α as described above. Oxygen levels have been suggested to directly modulate IKK expression and markedly suppress the induction of IL-1β mRNA expression via the LPS-mediated expression of NF-kappa-B. On the other hand, a previous study reported that FIH regulated the activity, but not the expression of HIF-1α (Ema et al., 1999). In the present study, in contrast to PHD, there was no direct interaction between FIH and IKKα/β. Furthermore, FIH has been shown to hydroxylate IκBα (Cockman et al., 2006); however, our results did not show these interactions, even with the chemiluminescence detection of immunoblotting. On the other hand, the activity of HIF-1α is regulated by Ref-1 (Ema et al., 1999), and the expression of Ref-1 is regulated by NF-kappa-B, particularly p65 (Kobayashi et al., 2021). In other words, ROS are generated under hypoxic conditions and reduce the expression of the Ref-1 protein (Kobayashi et al., 2021). Nevertheless, ROS produced under hypoxic conditions also activate NF-kappa-B. Therefore, the complex relationship between hypoxia and NF-kappa-B activity warrants further study.
ACKNOWLEDGMENT
This study was supported by the Japan Society for the Promotion of Science KAKENHI (Grant 17K08581 to SI).
Conflict of interest
The authors declare that there is no conflict of interest.
REFERENCES
- Cockman, M.E., Lancaster, D.E., Stolze, I.P., Hewitson, K.S., McDonough, M.A., Coleman, M.L., Coles, C.H., Yu, X., Hay, R.T., Ley, S.C., Pugh, C.W., Oldham, N.J., Masson, N., Schofield, C.J. and Ratcliffe, P.J. (2006): Posttranslational hydroxylation of ankyrin repeats in IkappaB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH). Proc. Natl. Acad. Sci. USA, 103, 14767-14772.
- Dames, S.A., Martinez-Yamout, M., De Guzman, R.N., Dyson, H.J. and Wright, P.E. (2002): Structural basis for Hif-1 alpha /CBP recognition in the cellular hypoxic response. Proc. Natl. Acad. Sci. USA, 99, 5271-5276.
- DiDonato, J.A., Hayakawa, M., Rothwarf, D.M., Zandi, E. and Karin, M. (1997): A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature, 388, 548-554.
- D’Ignazio, L. and Rocha, S. (2016): Hypoxia induced NF-kB. Cells, 8, 10.
- Ema, M., Hirota, K., Mimura, J., Abe, H., Yodoi, J., Sogawa, K., Poellinger, L. and Fujii-Kuriyama, Y. (1999): Molecular mechanisms of transcription activation by HLF and HIF1alpha in response to hypoxia: their stabilization and redox signal-induced interaction with CBP/p300. EMBO J., 18, 1905-1914.
- Freedman, S.J., Sun, Z.Y., Poy, F., Kung, A.L., Livingston, D.M., Wagner, G. and Eck, M.J. (2002): Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1 alpha. Proc. Natl. Acad. Sci. USA, 99, 5367-5372.
- Fukuda, R., Hirota, K., Fan, F., Jung, Y.D., Ellis, L.M. and Semenza, G.L. (2002): Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J. Biol. Chem., 277, 38205-38211.
- Gilmore, T.D. (2006): Introduction to NF-kappaB: players, pathways, perspectives. Oncogene, 25, 6680-6684.
- Hinz, M. and Scheidereit, C. (2014): The IκB kinase complex in NF-κB regulation and beyond. EMBO Rep., 15, 46-61.
- Hayden, M.S. and Ghosh, S. (2008): Shared principles in NF-kappaB signaling. Cell, 132, 344-362.
- Karin, M. (1999): How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene, 18, 6867-6874.
- Kobayashi, Y., Oguro, A. and Imaoka, S. (2021): Feedback of hypoxia-inducible factor-1alpha (HIF-1alpha) transcriptional activity via redox factor-1 (Ref-1) induction by reactive oxygen species (ROS). Free Radic. Res., 55, 154-164.
- Laughner, E., Taghavi, P., Chiles, K., Mahon, P.C. and Semenza, G.L. (2001): HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol. Cell. Biol., 21, 3995-4004.
- Lee, J.W., Bae, S.H., Jeong, J.W., Kim, S.H. and Kim, K.W. (2004): Hypoxia-inducible factor (HIF-1)alpha: its protein stability and biological functions. Exp. Mol. Med., 36, 1-12.
- Maxwell, P.H., Wiesener, M.S., Chang, G.W., Clifford, S.C., Vaux, E.C., Cockman, M.E., Wykoff, C.C., Pugh, C.W., Maher, E.R. and Ratcliffe, P.J. (1999): The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature, 399, 271-275.
- Maxwell, P.H., Pugh, C.W. and Ratcliffe, P.J. (2001): The pVHL-hIF-1 system. A key mediator of oxygen homeostasis. Adv. Exp. Med. Biol., 502, 365-376.
- Nakayama, K., Gazdoiu, S., Abraham, R., Pan, Z.-Q. and Ronai, Z. (2007): Hypoxia-induced assembly of prolyl hydroxylase PHD3 into complexes: implications for its activity and susceptibility for degradation by the E3 ligase Siah2. Biochem. J., 401, 217-226.
- Ohh, M., Park, C.W., Ivan, M., Hoffman, M.A., Kim, T.Y., Huang, L.E., Pavletich, N., Chau, V. and Kaelin, W.G. (2000): Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat. Cell Biol., 2, 423-427.
- Perkins, N.D. (2007): Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat. Rev. Mol. Cell Biol., 8, 49-62.
- Perkins, N.D. (2012): The diverse and complex roles of NF-κB subunits in cancer. Nat. Rev. Cancer, 12, 121-132.
- Prabhakar, N.R. and Semenza, G.L. (2015): Oxygen Sensing and Homeostasis. Physiology (Bethesda), 30, 340-348.
- Rius, J., Guma, M., Schachtrup, C., Akassoglou, K., Zinkernagel, A.S., Nizet, V., Johnson, R.S., Haddad, G.G. and Karin, M. (2008): NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature, 453, 807-811.
- Scheidereit, C. (2006): IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene, 25, 6685-6705.
- Schofield, C.J. and Ratcliffe, P.J. (2004): Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol., 5, 343-354.
- Semenza, G.L. (2001): HIF-1 and mechanisms of hypoxia sensing. Curr. Opin. Cell Biol., 13, 167-171.
- Siswanto, F.M., Okukawa, K., Tamura, A., Oguro, A. and Imaoka, S. (2023): Hydrogen peroxide activates APE1/Ref-1 via NF-κB and Parkin: a role in liver cancer resistance to oxidative stress. Free Radic. Res., 57, 223-238.
- Strowitzki, M.J., Cummins, E.P. and Taylor, C.T. (2019): Protein Hydroxylation by Hypoxia-Inducible Factor (HIF) Hydroxylases: unique or Ubiquitous? Cells, 8, 384. 10.3390/cells8050384
- Tarhonskaya, H., Chowdhury, R., Leung, I.K., Loik, N.D., McCullagh, J.S., Claridge, T.D., Schofield, C.J. and Flashman, E. (2014): Investigating the contribution of the active site environment to the slow reaction of hypoxia-inducible factor prolyl hydroxylase domain 2 with oxygen. Biochem. J., 463, 363-372.
- Tergaonkar, V. (2006): NFkappaB pathway: a good signaling paradigm and therapeutic target. Int. J. Biochem. Cell Biol., 38, 1647-1653.
- Xue, J., Li, X., Jiao, S., Wei, Y., Wu, G. and Fang, J. (2010): Prolyl hydroxylase-3 is down-regulated in colorectal cancer cells and inhibits IKKbeta independent of hydroxylase activity. Gastroenterology, 138, 606-615.
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/50_105-t1.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/50_105-t2.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/50_105-t3.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)