Abstract
Exposure to fine particulate matter (PM2.5) has been epidemiologically reported to worsen the prognosis of ischemic stroke; however, the details have not been investigated. One of the major toxic mechanisms of PM2.5 inhalation is oxidative stress, which is mediated by reactive oxygen species generated by PM2.5 components such as metals and polycyclic aromatic hydrocarbons. In this study, we examined the effects of long-term exposure to urban particulate matter, focusing on oxidative stress, on prognosis after ischemic stroke in mice. When mice were intranasally exposed for 28 days to an urban aerosol collected in Beijing, China (CRM28), microglial activation was observed in the cerebral cortex, indicating that CRM28 induced neuroinflammation. CRM28 exposure resulted in increased serum levels of brain natriuretic peptide and troponin I, suggesting that cardiac injury was elicited by CRM28. Lung inflammation was also observed following CRM28 exposure; however, systemic inflammation was not detected. Mice exposed to CRM28 showed an exacerbation of mortality after ischemic stroke induction compared with vehicle mice. A vitamin E-rich diet suppressed CRM28-induced lipid peroxidation in the heart and lungs but not in the brain. A vitamin E-rich diet also attenuated cardiac injury and lung inflammation induced by CRM28 exposure, whereas neuroinflammation was not affected. Mortality after ischemic stroke improved with the administration of a vitamin E-rich diet. Considering that systemic inflammation did not occur, cardiac injury induced by oxidative stress under exposure to urban particulate matter may be involved in increased mortality after ischemic stroke. Antioxidation under air pollution is fundamental for protection against ischemic stroke.
INTRODUCTION
Exposure to fine particulate matter (PM2.5) is well known to be a significant risk factor for various respiratory disorders. Upon inhalation, these particles penetrate deep into the lungs. These particles disrupt nasal epithelial cell metabolism, compromise the epithelial barrier, and alter inflammatory processes (Zaręba et al., 2024). These processes can contribute to chronic respiratory diseases, such as asthma and chronic obstructive pulmonary disease. In addition, exposure to PM2.5 is significantly associated with adverse cardiovascular outcomes, including mortality from atherosclerotic cardiovascular disease. Specific PM2.5 components such as metals and polycyclic aromatic hydrocarbons (PAHs) can increase the expression of cardiovascular biomarkers such as nitric oxide and adhesion molecules (Kahe et al., 2024).
Higher concentrations of PM2.5 have been reported to correlate with a greater incidence of cerebrovascular events, including ischemic strokes, in vulnerable populations such as postmenopausal women (Kulick et al., 2023) and even in the general population (Verhoeven et al., 2021). A 10 μg/m3 increase in PM2.5 was linked to a 1.84% rise in ischemic stroke mortality (From the American Association of Neurological Surgeons et al., 2018). Additionally, the Reasons for Geographic and Racial Differences in Stroke (REGARDS) study highlighted that the impact of PM2.5 on stroke risk varied by ecological region, indicating that environmental factors may influence these associations (Riggs et al., 2024). Overall, the evidence underscores the detrimental effects of PM2.5 exposure on stroke risk and outcomes.
PM2.5 can be transferred to the brain through two main pathways: passing through the lung-blood and blood-brain barriers or directly entering the brain tissue via the olfactory nerve (Li et al., 2022). PM2.5 exposure leads to the upregulation of pro-inflammatory proteins in the microglia, a type of immune cell in the brain, primarily through the Toll-like receptor 4/nuclear factor-κB pathway (Zhang et al., 2024). This process can be mediated by oxidative stress, as indicated by the increased production of reactive oxygen species (ROS). Treatment with antioxidants, such as acetylcysteine, can mitigate these effects, suggesting a role for oxidative stress in PM2.5-induced neuroinflammation. Recently, the chemokine receptor CCR5 was identified as a target of PM2.5 in the olfactory bulb, leading to microglial activation and subsequent neuroinflammation (Wei et al., 2024). Metals, including Mn and Pb, in PM2.5 were significant contributors to this process.
We previously reported that brain damage accompanied by ischemic stroke was potentiated by short-term exposure to urban particulate matter (7 days) and that ischemic inflammation was additionally enhanced by neuroinflammation accompanied by exposure to urban particulate matter (Tanaka et al., 2023). Importantly, respiratory and cardiovascular defects are reported to be able to contribute to cerebrovascular disorders, including strokes (Rojek et al., 2016; Xie et al., 2024), and it is considered that the longer the PM2.5 exposure is, the more severe this effect might become. In a study on the influence of PM2.5 exposure duration and concentration on outpatient visits of an urban hospital, while cardiovascular outpatient visits increased by exposure within less than 15 days, cardiovascular and cerebrovascular outpatient visits showed an overall upward trend by exposure with more than 30 days (Hu et al., 2023). Therefore, in this study, we aimed to examine the prognosis of cerebral infarction after long-term (28 days) exposure to urban particulate matter in mice, as well as to monitor the systemic effects of urban particulate matter to clarify the changes associated with worsening of the prognosis of cerebral ischemic stroke.
MATERIALS AND METHODS
Mice
All animal investigations were conducted according to Declaration of Helsinki principles. In addition, all animal procedures were performed in accordance with the Fundamental Guidelines for Proper Conduct of Animal Experiments and Related Activities in Academic Research Institutions under the Jurisdiction of the Ministry of Education, Culture, Sports, Science and Technology, Japan. The Animal Care and Use Committee of Hiroshima University approved the experimental protocols (No. C18-16-4 and C20-33).
Male ICR mice were purchased from CREA Japan (Tokyo, Japan). Four to five mice were housed in each cage. The animals were maintained on a 12:12 hr light/dark cycle and had free access to water and food. The mice were allowed to adapt to the facility for 1 week.
Mice received a normal diet (0.076 mg α-tocopherol/g diet) or a diet supplemented with vitamin E (VE) (1.342 mg α-tocopherol/g diet). The α-tocopherol was obtained from Wako Pure Industries Limited (Osaka, Japan). There were no differences in the dietary bulk among the experimental groups.
Particulate matter exposure
Urban aerosols collected from filters in a central ventilation system in a building in the Beijing City Center from 1996 to 2005 (CRM28) were purchased from the National Institute for Environmental Studies. CRM28 was sieved using a 32 μm sieve. The diameters of over 40% of CRM28 particles were less than 2 μm and 99% of the particles were less than 10 μm. The PM2.5 used in this study was collected in Yokohama, Japan, using a cyclone device equipped with an impactor as previously described (Ishihara et al., 2022a). Contents of PM2.5 collected in Yokohama, Japan were previously reported (Kono et al., 2024). PM2.5 included various shapes of nanoparticles confirmed by electron microscopy and atomic force microscopy (Fig. S1). CRM28 or PM2.5 were suspended in saline with sonication (10 W for 10 sec) at concentrations of 10 and 100 µg/10 µL. The resulting suspension of 10 µL was immediately applied into the nasal cavity of experimental mice using a micropipette. The vehicle mice were intranasally administered an equal volume of saline.
Microglial staining
Immunohistochemistry was performed as described previously (Ishihara et al., 2022b). The antibodies used in this study are listed in Table S1. The images were processed using the Zen image acquisition software package (Carl Zeiss, Oberkochen, Germany) and ImageJ software (National Institutes of Health, Bethesda, MD, USA). The soma area was evaluated using images of ionized calcium-binding adaptor molecule 1 (Iba1) staining, and the amoeboid score was calculated using the following formula: (soma area/entire Iba1-stained area) × 100 (%). The Iba1- and CD68-costained areas were analyzed to calculate the CD68-stained area.
Measurement of heart rate and blood pressure
Mice were put into the warmer, which was kept at 39°C, and the cuff pressure sensor was attached to the tail. Systolic, median, and diastolic blood pressure and heart rate were measured using a BP-98A sphygmomanometer (Bio Research Center Co. Ltd., Nagoya, Japan).
Assay for blood coagulation
Blood was collected from the inferior vena cava, and plasma was separated using sodium citrate. Coagulation assays were performed with a blood coagulation analyzer CA-101 (Sysmex Corporation, Kobe, Japan) using Thromborel S (Sysmex Corporation), Actin FSL (Sysmex Corporation), and Multifibern U (Sysmex Corporation) kits to measure prothrombin time, activated partial thromboplastin time, and fibrinogen concentration, respectively, according to the manufacturer’s instructions.
Determination of concentrations of serum B-type natriuretic peptide (BNP) and troponin I (TnI)
Blood was collected from the inferior vena cava and serum was prepared. Serum levels of BNP and TnI were measured using a RayBio Mouse BNP Enzyme Immunoassay Kit (RayBiotech, Peachtree Corners, GA, USA) and a Mouse Troponin I ELISA Kit (Abcam, Cambridge, UK), respectively, according to the manufacturer’s instructions.
Measurement of bronchoconstriction
Enhanced pause (PenH) was used as a parameter of bronchoconstriction in each conscious mouse as assessed by whole-body plethysmography (Emka Technologies, Sterling, VA, USA). The pressure signal results from pressure and volume changes in the chamber during the animal’s respiratory cycle. From these pressure signals, the phases of the respiratory cycle, tidal volumes, and the index of airway caliber PenH can be calculated. PenH is a dimensionless value that reflects changes in the waveform of the pressure signal resulting from both inspiration (PIP) and expiration (PEP), combined with a timing comparison of early and late expiration (pause).
Different concentrations of methacholine (MCh; 0, 3.1, 6.3, 12.5, and 25 mg/mL) were added to the chamber. The pressure signal was monitored for 3 min during each MCh inhalation. The average PenH during the latter 1 min of the 3 min was used as an index of bronchoconstriction for each MCh inhalation.
Total RNA extraction and real-time PCR
mRNA levels were determined according to the protocol described in our previous study (Ishihara et al., 2015). The primer sequences are listed in Table S2. mRNA levels were normalized to the level of β-actin, and the values of the treated samples were divided by those of the untreated samples to calculate the relative mRNA levels.
Periodic acid-Schiff (PAS) staining
The lungs were perfused with PBS and fixed for 1 d using 4% paraformaldehyde. A paraffin embedding block was prepared and cut into 10 μm sections with a microtome. Slices were stained using a PAS staining kit (Sigma-Aldrich, St. Louis, MO, USA). The stained images were captured using a BX61 upright microscope (Olympus Corporation, Tokyo, Japan).
Measurement of lipid peroxides
Thiobarbituric acid-reactive substance (TBARS) content was estimated using the method described in our previous reports (Ishihara et al., 2012; Ishihara et al., 2009) and was used as an index of lipid peroxidation. Briefly, the tissue was powdered using a Cryo Press (Microtec Company Limited, Chiba, Japan) at liquid nitrogen temperature and homogenized in 1.15% KCl solution. The homogenate was mixed with dodecyl sulfate, thiobarbituric acid, and dibutylhydroxytoluene in an acetic acid buffer (pH 3.5). The mixture was incubated at 100°C for 60 min, followed by extraction with a 1-butanol-pyridine solution. Absorbance of the extract was measured at 532 nm.
Preparation of the photothrombosis model
Cortical photothrombosis is induced by rose bengal injection and subsequent radiation (Tanaka et al., 2023). Anesthesia was initially achieved using 2.5% isoflurane, which was then reduced to 1.5% during the surgical procedure. Mice were placed in a stereotaxic frame and the scalp was incised. Rose bengal (10 mg/mL in sterile saline) was injected intraperitoneally at a dose of 50 mg/kg. An LED (170 mW, 561 nm, light diameter: 4 mm; M565L3, Thorlabs Inc. Newton, NJ, USA) was illuminated for 20 min, positioned posterior 2 mm and left 2 mm relative to the bregma (motor and somatosensory areas). Decreases in blood flow were determined using laser Doppler flowmetry (Unique Medical Co., Ltd., Tokyo, Japan), and mice with a 70–85% decrease in blood flow were used for subsequent experiments. The scalp was sutured and 1 mL of saline was injected subcutaneously.
Staining of the infarct region
The infarct size was assessed using 2,3,5-triphenyltetrazolium chloride (TTC) staining (Tanaka et al., 2018). The brains were sliced into 1-mm-thick coronal sections and stained with a 1% TTC solution in PBS at 37°C for 10 min. TTC-stained brain sections were analyzed using ImageJ software (National Institutes of Health).
Statistics
All data are presented as the mean ± standard deviation (S.D.). Statistical analyses were performed using GraphPad Prism 10 software (GraphPad Software, San Diego, CA, USA). Student's t-test, one-way analysis of variance (ANOVA) with Dunnett's corrected multiple comparison test, and two-way ANOVA with Tukey's corrected multiple comparison test were used to determine the significant differences between the means of two or more independent groups. Kaplan-Meier analysis was used for survival analysis, and survival differences between groups were assessed using the log-rank test. Statistical significance was set at p < 0.05. The results are described in the text or indicated in each figure.
RESULTS
Effects of long-term CRM28 exposure on physiological function and tissue damages
We have previously reported that short-term (7 day) exposure to urban particulate matter exacerbates decreases in motor function accompanied by ischemic stroke in mice (Tanaka et al., 2023). In this study, the effects of long-term exposure (28 days) to urban particulate matter on the prognosis of ischemic stroke were investigated, focusing on the peripheral effects of urban particulate matter and its action on the central nervous system (the experimental protocol is shown in Fig. S2A). The olfactory nerve is reportedly a pathway to carry PM2.5 to the brain (Liu et al., 2022). In addition, compared to respiratory exposure, intranasal exposure is less invasive and thus less damaging to animals. Therefore, we selected intranasal exposure of CRM28 and PM2.5 in this study.
While intranasal exposure to 10 μg CRM28 for 28 days did not affect microglial activity, 100 μg CRM28 significantly increased amoeboid score and CD68-stained area, indicating that microglial activation was induced by CRM28 exposure (Fig. 1). CRM28 exposure up to 100 μg did not alter cardiodynamics such as heart rate and blood pressure (Fig. 2A-D) and also did not alter blood coagulation status evaluated by 3 indices; prothrombin time, activated partial thromboplastin time, and fibrinogen concentration (Fig. 2E-G).

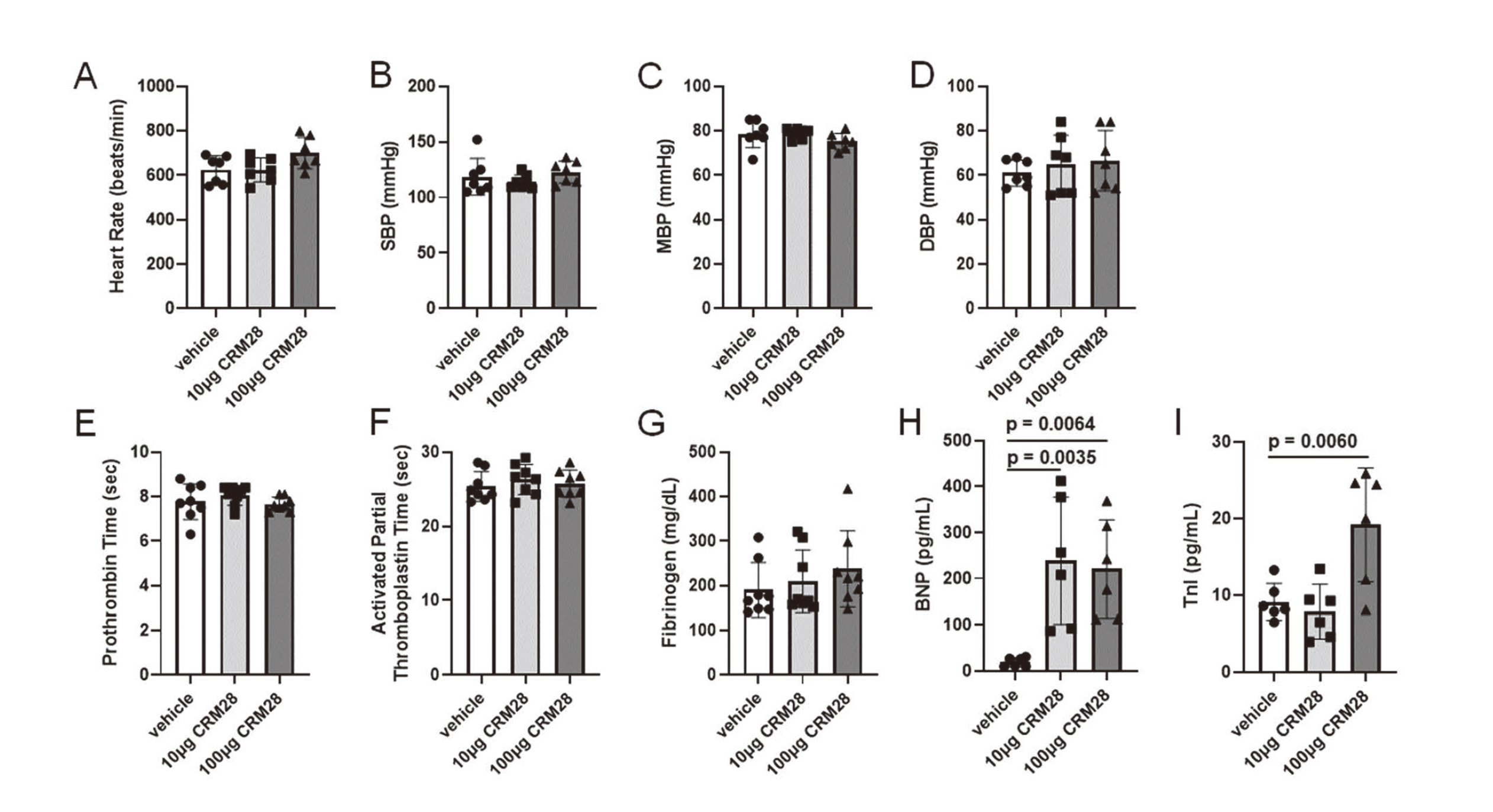
BNP is a crucial hormone produced by cardiomyocytes in the ventricular heart tissue in response to myocardial stretching and stress. Elevated BNP levels are clinically significant and can serve as biomarkers for the diagnosis and managing congestive heart failure (Kuwahara et al., 2018). TnI is a crucial protein in the troponin complex, primarily found in cardiac muscles, where it plays a vital role in regulating contraction and relaxation through calcium sensitivity. It also serves as a specific biomarker of myocardial injury, making it essential for clinical diagnostics, particularly in conditions such as myocardial infarction (Keller et al., 2009). When serum levels of BNP and TnI were measured, exposure to 10 μg CRM28 for 28 days significantly increased serum BNP concentration but not serum TnI levels (Fig. 2H and I). Both levels of BNP and TnI in the serum were significantly elevated by exposure to 100 μg CRM28 for 28 days (Fig. 2H and I). Short-term exposure of CRM28 did not affect the levels of BNP and TnI in serum (Fig. S3). These results suggested that long-term CRM28 exposure can cause cardiac damage.
Using barometric whole-body plethysmography and an increase in PenH as an index of airway obstruction, we measured the responses to inhaled MCh in CRM28-treated mice (Hamelmann et al., 1997). PenH increased in an MCh concentration-dependent manner in all groups (Fig. 3A). When the area under curve (AUC) was compared, although AUC of the 10 μg CRM28-treatment group did not show any difference from the AUC of vehicle group (p = 0.1177 analyzed using one-way ANOVA with Dunnett's corrected multiple comparison tests), the AUC of the 100 μg CRM28-treatment group was significantly larger than that of vehicle group (p = 0.090), indicating that exposure to 100 μg CRM28 for 28 days can induce airway hyperreactivity. Exposure to 10 μg CRM28 elicited cyclooxygenase-2 (COX-2) expression and 100 μg CRM28 exposure increased both interleukin-6 (IL-6) and COX-2 expression in the lung (Fig. 3B and C). No PAS-stained areas were detected in any of the three groups (Fig. 3D). Collectively, these results suggest that long-term exposure to CRM28 induces airway hyperreactivity and lung inflammation.

When we measured cytokine levels (IL-2, IL-4, IL-6, IL-17A, and tumor necrosis factor alpha [TNFα]) in the serum of vehicle, 10 μg and 100 μg CRM28-treated mice, there was no change in serum cytokine concentrations in the three groups (Fig. S4). Therefore, exposure to CRM28 for 28 days did not elicit systemic inflammation.
Effects of long-term CRM28 exposure on mortality after ischemic stroke
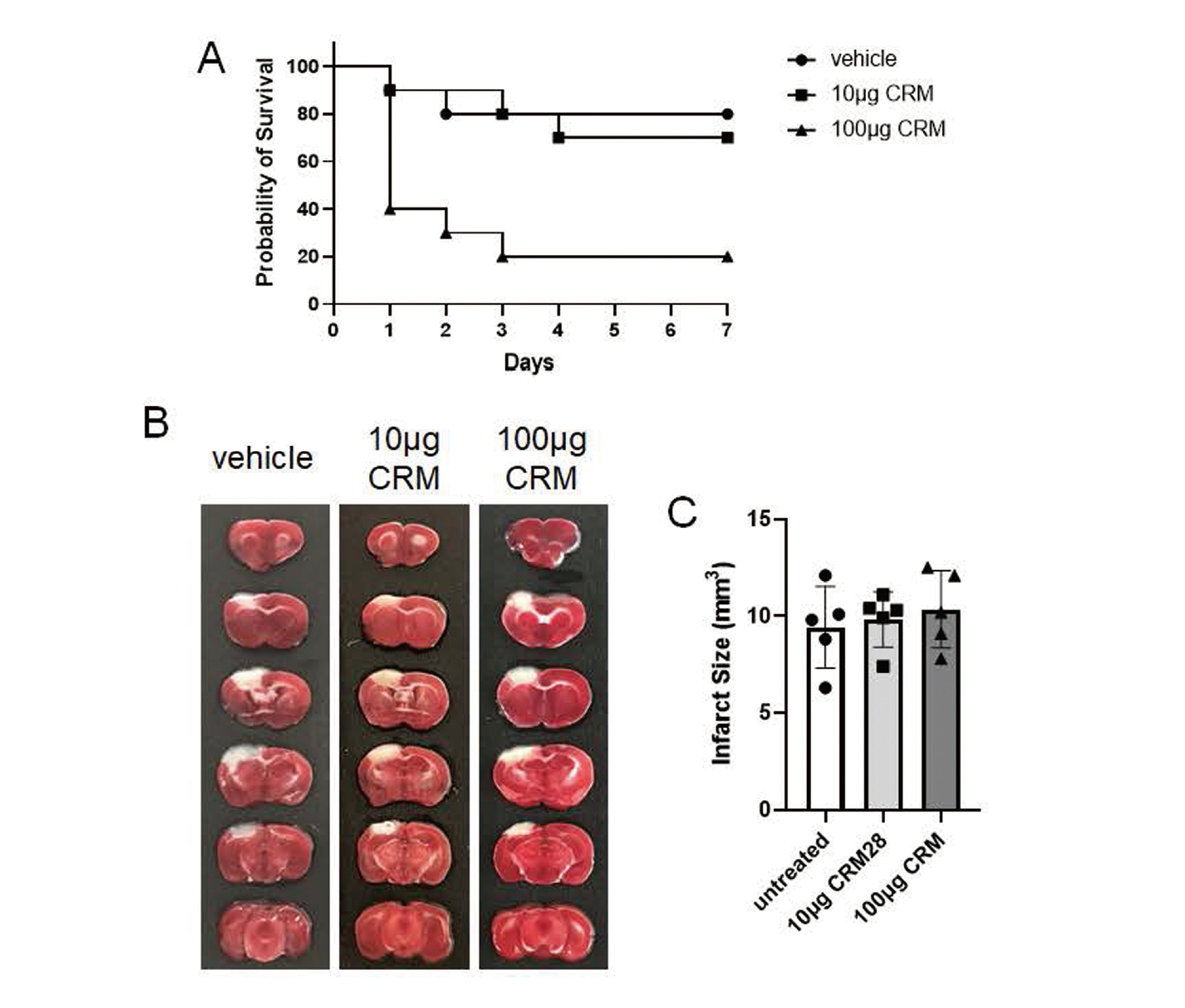
The photothrombosis model is widely used to induce localized ischemic injuries in the brain (Uzdensky, 2018). In the present study, ischemia was induced in an area spanning from the somatosensory cortex to the motor cortex. In vehicle- and 10 μg CRM28-treated groups, almost all mice were alive for 7 days after induction of ischemia (Fig. 4A). On the other hand, the mortality of mice exposed to 100 μg CRM28 significantly decreased compared with that of the vehicle mice (p = 0.0069, calculated using log-rank test). There was no difference in infarct volume among the three groups (Fig. 4B and C).
Effects of VE supplementation on oxidative stress, inflammation, and lung dysfunction induced by CRM28 exposure
Oxidative stress is a major mechanism underlying the harmful effects of PM2.5 exposure. Chronic exposure to PM2.5, in which oxidative stress exacerbates airway inflammation and hyperresponsiveness, is associated with respiratory diseases, including asthma (Liu et al., 2022). Cardiovascular diseases are also linked to exposure to PM2.5, and oxidative stress has been identified as a key pathway in the development of these conditions (Bhatnagar, 2022). Therefore, we examined the effects of antioxidant VE on CRM28-induced tissue damage and the prognosis of ischemic stroke. VE is a natural and potent antioxidant with lipophilicity, and thus can be maintained in the body for a long time. We have reported that oxidative stress in the hippocampus of Alzheimer's disease model mice was suppressed by feeding a diet supplemented with VE (Ishihara et al., 2013).
We prepared a VE-rich diet and fed it to mice in combination with vehicle or 100 μg CRM28 exposure for 28 days. The experimental protocol is shown in Fig. S2B. Intranasal exposure to 100 μg CRM28 for 28 days increased the TBARS levels in the heart and lungs, but not in the cerebral cortex (Fig. 5). VE supplementation significantly suppressed the increase in TBARS levels in the heart and lungs (Fig. 5), indicating that CRM28 exposure induced oxidative stress in these tissues. VE supplementation did not affect the increased amoeboid score or CD68-stained area induced by CRM28 exposure (Fig. 6A and B). Administration of VE significantly reduced the serum BNP and TnI levels that were elevated by CRM28 exposure (Fig. 6C and D), indicating that oxidative stress elicited by CRM28 is involved in myocardial injury. PenH values increased in an MCh concentration-dependent manner in all the groups examined (Fig. 6E). The AUC of 100 μg CRM28-treatment group significantly larger than that of vehicle group (p < 0.0001 analyzed using two-way ANOVA with Tukey's corrected multiple comparison tests). VE supplementation largely suppressed the increase in the AUC after CRM28 exposure (p = 0.0007). VE administration clearly attenuated the increased expression of IL-6 and COX-2 induced by CRM28 exposure (Fig. 6F and G). Taken together, these results suggest that airway hyper-reactivity, lung inflammation, and cardiac injury are elicited by oxidative stress accompanied by long-term CRM28 exposure.

We next compared mortality after ischemic stroke between the normal and VE-rich diet groups. After photothrombosis, the mortality of mice with normal diet exposed to 100 μg CRM28 significantly decreased compared with that of vehicle mice (Fig. 7, p = 0.0009, calculated using log-rank test). VE supplementation clearly attenuated increased mortality associated with CRM28 exposure (Fig. 7, p = 0.0405). Therefore, the oxidative stress induced by CRM28 exposure is thought to contribute to mortality after ischemic stroke.
Effects of long-term exposure to PM2.5 on mortality after ischemic stroke
Next, to confirm the generality of the increased mortality after cerebral ischemic stroke due to long-term exposure to PM2.5, we challenged PM2.5 collected in the outdoor environment by a cyclone. When mice were exposed to PM2.5 collected in Yokohama, Japan from June to September, 2021 (shown as PM2.5 Yokohama 2021) at a dose of 100 μg/day/mouse for 28 days, mortality after photothrombosis significantly increased (Fig. 8, p = 0.0082). PM2.5 collected in Yokohama, Japan, from November 2021 to February 2022 (shown as PM2.5 Yokohama 2022) also increased mortality after ischemia (Fig. 8, p = 0.0275). Therefore, long-term exposure to PM2.5, collected in Yokohama, Japan, exacerbates mortality after ischemic stroke.
DISCUSSION
Exposure to CRM28 induced neuroinflammation, cardiac injury, and lung inflammation, but did not affect blood coagulation or systemic inflammation, suggesting that inhaled PM affected the brain, heart, and lungs without systemic inflammation. Oberdorster et al. reported that inhaled carbon particles were distributed in the brain (Oberdörster et al., 2004). PM2.5 is taken up by olfactory neurons, reaches the olfactory bulb via axonal transport, and is spread throughout the CNS via the intraneural spaces. The lungs are a direct PM2.5 target, and inhaled PM2.5 passes through the respiratory tract to reach the lungs. However, there is a controversy regarding how PM2.5 is transferred to the heart. PM2.5 exposure induces oxidative stress, which disrupts the redox balance in cardiac tissues, leading to mitochondrial dysfunction and inflammation (Sivakumar and Kurian, 2024). PM2.5 also activates inflammatory pathways, contributing to endothelial dysfunction and atherosclerosis (Krittanawong et al., 2023). Because long-term exposure to CRM28 did not elicit systemic inflammation in this study, systemic inflammation is unlikely to be involved in cardiac injury. CRM28 contains a large number of polycyclic aromatic hydrocarbons (PAHs) (Tanaka et al., 2023) that are known to act as agonists of the aryl hydrocarbon receptor (AhR). It has been reported that PM2.5 induces cardiac defects via AhR-mediated mitochondrial damage (Chen et al., 2024). Thus, PAHs entering the blood might act on cardiac AhRs to induce mitochondrial damage. Cardiac mitochondrial dysfunction is particularly pronounced in interfibrillar mitochondria (IFM), which show elevated oxidative stress (Sivakumar et al., 2022). Our data showed that long-term CRM28 exposure induced lipid peroxidation in the heart. Therefore, these findings support the hypothesis that PAHs in CRM28 affect cardiac oxidative injury.
In this study, we administered CRM28 intranasally at 10 and 100 µg/mouse/day as in our previous report (Tanaka et al., 2023). Converting the exposure in this study to the exposure per body weight for 28 days, 10 µg/mouse/day is 7.0 mg/kg if mouse weight is 40 g for calculation. In Japan, the environmental standard for PM2.5 is "the daily average value of PM2.5 is 35 μg/m3 or less". The ventilation volume of a person with a body weight of 60 kg is calculated to be 0.5 L per breath, thus approximately 14 m3 per day. Then, it became 8.4 µg/kg, as calculated by the Japanese environmental standard. Regarding this calculation, exposure for 28 days at 10 or 100 µg/mouse/day is equivalent to exposure for approximately 2.3 or 23 years, respectively, at the maximum value within the environmental standards in Japan. Air pollution has regional and seasonal characteristics and there are many areas where PM2.5 in the atmosphere is relatively high; PM2.5 in northern India recently increased to approximately 500 µg/m3 in November (Singh et al., 2023). Therefore, the effect of PM2.5 on the nervous system may not be negligible in such areas with high PM2.5 concentrations.
Oxidative stress is defined as the balance between ROS production and antioxidant levels, such as those of antioxidative enzymes distributed in tissues. Therefore, not only the amount of ROS but also the antioxidant capacity of tissues are important factors in determining susceptibility to oxidative stress. In this study, oxidative stress was significantly induced in the lungs and myocardium, but slightly in the brain, following CRM28 exposure. The antioxidant capacities of the lungs and heart are much lower than those of the liver and kidneys (Cao et al., 1996). The brain consumes 20% of the oxygen used throughout the body and is thus exposed to large amounts of ROS. Therefore, the brain is believed to exhibit high antioxidant activity. Although the olfactory epithelium delivers PM2.5 and its components to the brain (Oberdörster et al., 2004), it is presumed that the amount transported is not particularly high. These may explain the degree of oxidative stress in different tissues upon exposure to CRM28.
In our previous study, motor dysfunction accompanied by ischemic stroke was potentiated by short-term (7 day) urban particulate matter exposure (Tanaka et al., 2023). However, short-term exposure did not affect the mortality after ischemic stroke. In this study, we induced cerebral ischemia after 28 days of CRM28 or PM2.5 exposure and then mortality in the acute phase was significantly increased following urban particulate matter exposure. Therefore, it has been suggested that long-term urban particulate matter exposure affects survival rates after cerebral ischemic stroke. Epidemiological studies have reported that air pollution increases mortality one year after the onset of cerebral stroke (Chen et al., 2019). Our study supports this epidemiological finding, and a correlation between PM2.5 exposure and survival rate after cerebral stroke may occur in highly polluted areas. Importantly, while short-term CRM28 exposure did not increase serum levels of BNP and TnI, the amount of BNP and TnI in serum was significantly elevated by long-term CRM28 exposure significantly. As described above, BNP and TnI are specific biomarkers for myocardial injury. Therefore, the outcome of cerebral ischemic stroke may be determined by whether or not exposure to PM2.5 causes myocardial defect.
Supplementation with dietary VE significantly suppressed myocardial injury and respiratory inflammation, and improved mortality after cerebral ischemia. Therefore, myocardial damage and respiratory inflammation may correlate with mortality after ischemic stroke. Cardiovascular complications are the leading causes of post-stroke mortality. van der Bilt et al. (2009) revealed that cardiac dysfunction is associated with poor outcomes after subarachnoid hemorrhage (van der Bilt et al., 2009). Ventricular dysfunction is prevalent in stroke patients and is correlated with higher stroke severity and mortality (Battaglini et al., 2020). This underscores the importance of cardiac evaluation and intervention in the management of patients with stroke. The interaction between cardiac function and arterial health, particularly in the presence of hypertension and atherosclerosis, further complicates the clinical outcome. Reduced cardiac output and increased vascular stiffness can lead to cerebral hypoperfusion, worsened neurological outcomes, and increased mortality risk (Rojek et al., 2016). Exposure to PM2.5, which induces abnormal lipid metabolism and causes arteriosclerosis (Zhao et al., 2023), also increases the incidence of cerebral ischemic stroke (Toubasi and Al-Sayegh, 2023; Lu et al., 2023). Although cardiac damage is highly related to the prognosis of stroke, there was no change in blood pressure or heart rate between the vehicle- and CRM28-treated mice in this study. Therefore, a detailed analysis of the hearts of mice exposed to CRM28 is required. Inflammatory responses in the lungs contribute to the overall inflammatory cascade following ischemic stroke (Xie et al., 2024). This interaction, through the circulation between the lungs and brain, plays a crucial role in mediating and regulating post-stroke inflammation. However, as discussed above, since systemic inflammation was not detected in mice exposed to CRM28 in this study, there is a lower possibility that inflammation is transferred to the brain.
In conclusion, we have previously shown that short-term exposure only resulted in a deterioration of motor function (Tanaka et al., 2023), and we revealed in this study that long-term urban particulate matter exposure was involved in increased mortality after ischemic stroke. Therefore, the longer the exposure duration to urban particulate matter was, the more severe the prognosis became. The World Health Organization (WHO) states that almost the entire global population (99%) breathes air that exceeds the WHO air quality limits; thus, these people are expected to be at a high risk of worsen prognosis after ischemic stroke. Reducing air pollution should be one measure, but it is also important to uncover the mechanism by which PM2.5 worsens the prognosis of cerebral infarction in order to develop mechanism-based prevention and treatment approaches. Cardiac oxidative injury induced by long-term PM exposure is likely to contribute to a worse prognosis after stroke. Taken together, the findings of this study indicate that antioxidation under air pollution conditions is fundamental for protection against ischemic stroke.
ACKNOWLEDGMENTS
This study was supported by KAKENHI grants from the Japan Society for the Promotion of Science (grant number 24K03068 to TO and 24K03085 to YI), the Environment Research and Technology Development Funds of the Environmental Restoration and Conservation Agency of Japan (JPMEERF20205007 and JPMEERF20245004 to YI) and the Smoking Research Foundation (to YI).
Conflict of interest
The authors declare that there is no conflict of interest.
REFERENCES
- Battaglini, D., Robba, C., Lopes da Silva, A., Dos Santos Samary, C., Leme Silva, P., Dal Pizzol, F., Pelosi, P. and Rocco, P.R. (2020): Brain-heart interaction after acute ischemic stroke. Crit. Care, 24, 163.
- Bhatnagar, A. (2022): Cardiovascular Effects of Particulate Air Pollution. Annu. Rev. Med., 73, 393-406.
- Cao, G., Giovanoni, M. and Prior, R.L. (1996): Antioxidant capacity in different tissues of young and old rats. Proc. Soc. Exp. Biol. Med., 211, 359-365.
- Chen, G., Wang, A., Li, S., Zhao, X., Wang, Y., Li, H., Meng, X., Knibbs, L.D., Bell, M.L., Abramson, M.J., Wang, Y. and Guo, Y. (2019): Long-Term Exposure to Air Pollution and Survival After Ischemic Stroke. Stroke, 50, 563-570.
- Chen, J., Zhang, M., Aniagu, S., Jiang, Y. and Chen, T. (2024): PM2.5 induces cardiac defects via AHR-SIRT1-PGC-1α mediated mitochondrial damage. Environ. Toxicol. Pharmacol., 106, 104393.
- David Sacks, D., Baxter, B., Campbell, B.C., et al. (2018): Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke, 13, 612-632.
- Hamelmann, E., Schwarze, J., Takeda, K., Oshiba, A., Larsen, G.L., Irvin, C.G. and Gelfand, E.W. (1997): Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am. J. Respir. Crit. Care Med., 156, 766-775.
- Hu, J., Wang, F. and Shen, H. (2023): The influence of PM2.5 exposure duration and concentration on outpatient visits of urban hospital in a typical heavy industrial city. Environ. Sci. Pollut. Res. Int., 30, 115098-115110.
- Ishihara, N., Okuda, T., Hagino, H., Oguro, A., Tani, Y., Okochi, H., Tokoro, C., Fujii-Kuriyama, Y., Itoh, K., Vogel, C.F. and Ishihara, Y. (2022a): Involvement of polycyclic aromatic hydrocarbons and endotoxin in macrophage expression of interleukin-33 induced by exposure to particulate matter. J. Toxicol. Sci., 47, 201-210.
- Ishihara, Y., Honda, T., Ishihara, N., Namba, K., Taketoshi, M., Tominaga, Y., Tsuji, M., Vogel, C.F., Yamazaki, T., Itoh, K. and Tominaga, T. (2022b): A CCR5 antagonist, maraviroc, alleviates neural circuit dysfunction and behavioral disorders induced by prenatal valproate exposure. J. Neuroinflammation, 19, 195.
- Ishihara, Y., Itoh, K., Mitsuda, Y., Shimada, T., Kubota, T., Kato, C., Song, S.Y., Kobayashi, Y., Mori-Yasumoto, K., Sekita, S., Kirino, Y., Yamazaki, T. and Shimamoto, N. (2013): Involvement of brain oxidation in the cognitive impairment in a triple transgenic mouse model of Alzheimer’s disease: noninvasive measurement of the brain redox state by magnetic resonance imaging. Free Radic. Res., 47, 731-739.
- Ishihara, Y., Kawami, T., Ishida, A. and Yamazaki, T. (2012): Tributyltin induces oxidative stress and neuronal injury by inhibiting glutathione S-transferase in rat organotypic hippocampal slice cultures. Neurochem. Int., 60, 782-790.
- Ishihara, Y., Sekine, M., Nakazawa, M. and Shimamoto, N. (2009): Suppression of myocardial ischemia-reperfusion injury by inhibitors of cytochrome P450 in rats. Eur. J. Pharmacol., 611, 64-71.
- Ishihara, Y., Takemoto, T., Itoh, K., Ishida, A. and Yamazaki, T. (2015): Dual role of superoxide dismutase 2 induced in activated microglia: oxidative stress tolerance and convergence of inflammatory responses. J. Biol. Chem., 290, 22805-22817.
- Kahe, D., Sabeti, Z., Sarbakhsh, P., Shakerkhatibi, M., Gholampour, A., Goudarzi, G., Sharbafi, J., Dastgiri, S., Separham, A. and Seyedrezazadeh, E. (2024): Effect of PM2.5 exposure on adhesion molecules and systemic nitric oxide in healthy adults: the role of metals, PAHs, and oxidative potential. Chemosphere, 354, 141631.
- Keller, T., Zeller, T., Peetz, D., Tzikas, S., Roth, A., Czyz, E., Bickel, C., Baldus, S., Warnholtz, A., Fröhlich, M., Sinning, C.R., Eleftheriadis, M.S., Wild, P.S., Schnabel, R.B., Lubos, E., Jachmann, N., Genth-Zotz, S., Post, F., Nicaud, V., Tiret, L., Lackner, K.J., Münzel, T.F. and Blankenberg, S. (2009): Sensitive troponin I assay in early diagnosis of acute myocardial infarction. N. Engl. J. Med., 361, 868-877.
- Kono, M., Takaishi, M., Okuda, T., Fujihara, M., Noguchi, S. and Ishihara, Y. (2024): A simple air-liquid interface exposure system for exposing cultured human 3D epidermis and cornea to PM2.5 collected through cyclonic separation. J. Toxicol. Sci., 49, 61-68.
- Krittanawong, C., Qadeer, Y.K., Hayes, R.B., Wang, Z., Thurston, G.D., Virani, S. and Lavie, C.J. (2023): PM2.5 and cardiovascular diseases: State-of-the-Art review. Int. J. Cardiol. Cardiovasc. Risk Prev., 19, 200217.
- Kulick, E.R., Eliot, M.N., Szpiro, A.A., Coull, B.A., Tinker, L.F., Eaton, C.B., Whitsel, E.A., Stewart, J.D., Kaufman, J.D. and Wellenius, G.A. (2023): Long-term exposure to ambient particulate matter and stroke etiology: Results from the Women’s Health Initiative. Environ. Res., 224, 115519.
- Kuwahara, K., Nakagawa, Y. and Nishikimi, T. (2018): Cutting Edge of Brain Natriuretic Peptide (BNP) Research - The Diversity of BNP Immunoreactivity and Its Clinical Relevance. Circ. J., 82, 2455-2461.
- Li, W., Lin, G., Xiao, Z., Zhang, Y., Li, B., Zhou, Y., Ma, Y. and Chai, E. (2022): A review of respirable fine particulate matter (PM2.5)-induced brain damage. Front. Mol. Neurosci., 15, 967174.
- Liu, K., Hua, S. and Song, L. (2022): PM2.5 Exposure and Asthma Development: The Key Role of Oxidative Stress. Oxid. Med. Cell. Longev., 2022, 3618806.
- Lu, H., Wang, R., Li, J., Tong, M., Cao, M., Liu, H., Xiao, Q., Zheng, Y., Liu, Y., Guan, T. and Xue, T. (2023): Long-term exposure to the components of fine particulate matters and disability after stroke: Findings from the China National Stroke Screening Surveys. J. Hazard. Mater., 460, 132244.
- Oberdörster, G., Sharp, Z., Atudorei, V., Elder, A., Gelein, R., Kreyling, W. and Cox, C. (2004): Translocation of inhaled ultrafine particles to the brain. Inhal. Toxicol., 16, 437-445.
- Riggs, D.W., Baumgartner, K.B., Baumgartner, R., Boone, S., Judd, S.E. and Bhatnagar, A. (2024): Long-term exposure to air pollution and risk of stroke by ecoregions: the REGARDS study. Environ. Pollut., 345, 123367.
- Rojek, A., Gąsecki, D., Fijałkowski, M., Kowalczyk, K., Kwarciany, M., Wolf, J., Nyka, W., Boutouyrie, P., Laurent, S. and Narkiewicz, K. (2016): Left ventricular ejection fraction and aortic stiffness are independent predictors of neurological outcome in acute ischemic stroke. J. Hypertens., 34, 2441-2448.
- Singh, T., Matsumi, Y., Nakayama, T., Hayashida, S., Patra, P.K., Yasutomi, N., Kajino, M., Yamaji, K., Khatri, P., Takigawa, M., Araki, H., Kurogi, Y., Kuji, M., Muramatsu, K., Imasu, R., Ananda, A., Arbain, A.A., Ravindra, K., Bhardwaj, S., Kumar, S., Mor, S., Dhaka, S.K., Dimri, A.P., Sharma, A., Singh, N., Bhatti, M.S., Yadav, R., Vatta, K. and Mor, S. (2023): Very high particulate pollution over northwest India captured by a high-density in situ sensor network. Sci. Rep., 13, 13201.
- Sivakumar, B., AlAsmari, A.F., Ali, N., Waseem, M. and Kurian, G.A. (2022): Consequential Impact of Particulate Matter Linked Inter-Fibrillar Mitochondrial Dysfunction in Rat Myocardium Subjected to Ischemia Reperfusion Injury. Biology (Basel), 11.
- Sivakumar, B. and Kurian, G.A. (2024): PM2.5 toxicity in blood impairs cardiac redox balance and promotes mitochondrial dysfunction in rat heart that further aggravates ischemia reperfusion injury by modulating PI3K/AKT/mTOR/NF-kB signaling axis. J. Biochem. Mol. Toxicol., 38, e23718.
- Tanaka, M., Ishihara, Y., Mizuno, S., Ishida, A., Vogel, C.F., Tsuji, M., Yamazaki, T. and Itoh, K. (2018): Progression of vasogenic edema induced by activated microglia under permanent middle cerebral artery occlusion. Biochem. Biophys. Res. Commun., 496, 582-587.
- Tanaka, M., Okuda, T., Itoh, K., Ishihara, N., Oguro, A., Fujii-Kuriyama, Y., Nabetani, Y., Yamamoto, M., Vogel, C.F. and Ishihara, Y. (2023): Polycyclic aromatic hydrocarbons in urban particle matter exacerbate movement disorder after ischemic stroke via potentiation of neuroinflammation. Part. Fibre Toxicol., 20, 6.
- Toubasi, A. and Al-Sayegh, T.N. (2023): Short-term Exposure to Air Pollution and Ischemic Stroke: A Systematic Review and Meta-analysis. Neurology, 101, e1922-e1932.
- Uzdensky, A.B. (2018): Photothrombotic Stroke as a Model of Ischemic Stroke. Transl. Stroke Res., 9, 437-451.
- van der Bilt, I.A., Hasan, D., Vandertop, W.P., Wilde, A.A., Algra, A., Visser, F.C. and Rinkel, G.J. (2009): Impact of cardiac complications on outcome after aneurysmal subarachnoid hemorrhage: a meta-analysis. Neurology, 72, 635-642.
- Verhoeven, J.I., Allach, Y., Vaartjes, I.C., Klijn, C.J. and de Leeuw, F.E. (2021): Ambient air pollution and the risk of ischaemic and haemorrhagic stroke. Lancet Planet. Health, 5, e542-e552.
- Wei, S., Xu, T., Sang, N., Yue, H., Chen, Y., Jiang, T., Jiang, T. and Yin, D. (2024): Mixed Metal Components in PM2.5 Contribute to Chemokine Receptor CCR5-Mediated Neuroinflammation and Neuropathological Changes in the Mouse Olfactory Bulb. Environ. Sci. Technol., 58, 4914-4925.
- Xie, L., He, M., Ying, C. and Chu, H. (2024): Mechanisms of inflammation after ischemic stroke in brain-peripheral crosstalk. Front. Mol. Neurosci., 17, 1400808.
- Zaręba, Ł., Piszczatowska, K., Dżaman, K., Soroczynska, K., Motamedi, P., Szczepański, M.J. and Ludwig, N. (2024): The Relationship between Fine Particle Matter (PM2.5) Exposure and Upper Respiratory Tract Diseases. J. Pers. Med., 14.
- Zhang, L., Xu, F., Yang, Y., Yang, L., Wu, Q., Sun, H., An, Z., Li, J., Wu, H., Song, J. and Wu, W. (2024): PM2.5 exposure upregulates pro-inflammatory protein expression in human microglial cells via oxidant stress and TLR4/NF-κB pathway. Ecotoxicol. Environ. Saf., 277, 116386.
- Zhao, T., Li, X., Qian, H., Miao, X., Zhu, Y., Wang, J., Hui, J., Zhou, L. and Ye, L. (2023): PM2.5 induces the abnormal lipid metabolism and leads to atherosclerosis via Notch signaling pathway in rats. Toxicology, 485, 153415.