Abstract
SARS-CoV-2 whole-genome sequencing of samples from COVID-19 patients is useful for
informing infection control. Datasets of these genomes assembled from multiple hospitals
can give critical clues to regional or national trends in infection. Herein, we report a
lineage summary based on data collected from hospitals located in the Tokyo metropolitan
area. We performed SARS-CoV-2 whole-genome sequencing of specimens from 198 patients with
COVID-19 at 13 collaborating hospitals located in the Kanto region. Phylogenetic analysis
and fingerprinting of the nucleotide substitutions were performed to differentiate and
classify the viral lineages. More than 90% of the identified strains belonged to Clade
20B, which has been prevalent in European countries since March 2020. Only two lineages
(B.1.1.284 and B.1.1.214) were found to be predominant in Japan. However, one sample from
a COVID-19 patient admitted to a hospital in the Kanto region in November 2020 belonged to
the B.1.346 lineage of Clade 20C, which has been prevalent in the western United States
since November 2020. The patient had no history of overseas travel or any known contact
with anyone who had travelled abroad. Consequently, the Clade 20C strain belonging to the
B.1.346 lineage appeared likely to have been imported from the western United States to
Japan across the strict quarantine barrier. B.1.1.284 and B.1.1.214 lineages were found to
be predominant in the Kanto region, but a single case of the B.1.346 lineage of clade 20C,
probably imported from the western United States, was also identified. These results
illustrate that a decentralized network of hospitals offers significant advantages as a
highly responsive system for monitoring regional molecular epidemiologic trends.
Introduction
SARS-CoV-2 whole-genome sequencing of samples from COVID-19 patients is useful for
informing infection control.1 When multiple
COVID-19-positive cases occur simultaneously in a hospital, it is critical to determine
whether newly diagnosed patients have nosocomial infection or community infection. For
nosocomial infection, a thorough contact tracing of healthcare workers and inpatients is
essential, whereas, in the case of community infection, such persistent intrahospital
surveillance may not be necessary.
Datasets of whole viral genomes assembled from multiple hospitals can give critical clues
to regional or national trends. The national surveillance system developed in the United
Kingdom clarified that more than 1000 lineages had spread during the pre-lockdown period of
high travel volumes and few restrictions on international travel.2 Recently, genomic surveillance systems have successfully
identified two variants of concern, i.e., 501Y.V1 (B.1.1.7 originating in the UK) and
501Y.V2 (B.1.351 originating in South Africa), both of which spread rather
quickly.3,4 In Japan, five laboratories (including our
group) have deposited whole viral genome data in the public sequence database GISAID
(https://www.gisaid.org/): a total of 9943 sequences have been deposited, and some results
of national surveys have been summarized.5,6 These
studies indicated the importance of SARS-CoV-2 genome sequencing analysis for the prevention
of outbreaks during the early pandemic period (March to April 2020). SARS-CoV-2 whole-genome
data was generated using specimens from 198 patients with COVID-19 admitted to 13
collaborating hospitals located in the Tokyo metropolitan area. Data from the specimens were
accumulated and utilized for infection control and showed that the predominant lineages of
the SARS-CoV-2 strains were limited to only two closely related strains, B.1.1.284 and
B.1.1.214. This finding indicates that the quarantine system has been relatively successful.
However, one strain derived from a patient with nosocomial COVID-19 infection belonged to
the B.1.346 lineage, which has been prevalent in the western United States of America (USA)
from winter 2020. We suspect that this episode is attributable to undetected imported
infections from the USA.
Materials and Methods
Study design and patients
The study protocol was approved by the Institutional Review Board of Keio University
School of Medicine (approval number: 20200062) in association with each collaborating
hospitals’ review board and was conducted according to the principles of the Declaration
of Helsinki.
DNA sequencing method
Whole viral genome sequences were determined as described previously.1 Polymerase chain reaction-based amplification
was performed using Artic ncov-2019 primers in addition to a number 72-mutant primer,
version 3
(https://github.com/artic-network/artic-ncov2019/blob/master/primer_schemes/nCoV-2019/V3/nCoV-2019.tsv)
in two multiplex reactions according to the globally accepted nCoV-2019 sequencing
protocol (https://www.protocols.io/view/ncov-2019-sequencingprotocol-bbmuik6w). The
sequencing library for amplicon sequencing was prepared using the Next Ultra II DNA
Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA, USA). Paired-end
sequencing was performed on the MiSeq platform (Illumina, San Diego, CA, USA). The
bioinformatic pipeline used in this study, the mutation calling pipeline for
amplicon-based sequencing of the SARS-CoV-2 viral genome, is available at
https://cmg.med.keio.ac.jp/sars-cov-2/. All single nucleotide substitutions, including
non-synonymous and synonymous mutations, were annotated using ANNOVAR software and were
assessed using VarSifter (https://research.nhgri.nih.gov/software/VarSifter/). The
analytic protocol was described previously in our publication submitted on November 24,
2020, and is available on the preprint server medRxiv.7
Genetic clade or lineage naming in phylogenic tree analyses
Phylogenetic tree analysis was performed locally using the Augur program available from
Nextstrain (https://nextstrain.org/) and genome sequence data obtained in the current
study as well as data available from the global database EpiCov hosted at
GISAID.8 Nextclade
(https://clades.nextstrain.org/) was also used to generate fingerprinting patterns to
visualize the SARS-CoV-2 sequence alignments and similarities/identities among
samples.
We used the international genetic clade nomenclature system defined by Nextstrain.org
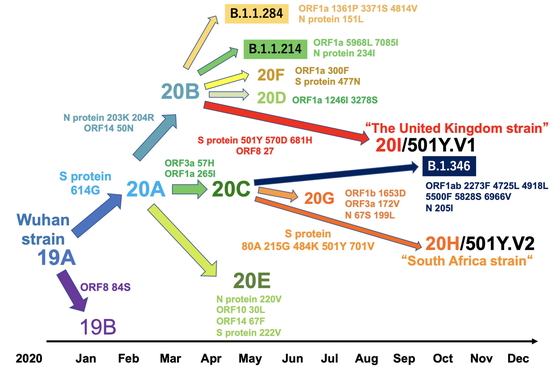
(https://virological.org/t/updated-nextstain-sars-cov-2-clade-naming-strategy/581) (Fig. 1). We also used the software Phylogenetic
Assignment of Named Global Outbreak Lineages (Pangolin;
https://cov-lineages.org/index.html) to assign viral lineages in an automatic and precise
manner.9
Results
Whole viral genome sequencing was carried out on samples from 198 COVID-19 patients
collected between March 2020 and November 2020. In total, 189 (95.5%) of the viral genomes
belonged to Clade 20B of the Clade system. Ninety-eight of 189 (51.9%) were classified as
B.1.1.284 (referred to as Clade 20B-T in our previous publication7) whereas 60 (31.7%) were classified as B.1.1.214. These two
lineages differ by six nucleotide substitutions. The total number of samples in the GISAID
database corresponding to B.1.1.284 and B.1.1.214 are 4183 and 1070, respectively, as has
been previously reported.7 The vast
majority of B.1.1.284 and B.1.1.214 cases were found in Japan. Outside Japan, B.1.1.284 and
B.1.1.214 are extremely rare: only eight such B.1.1.284 samples have been detected (two in
Thailand, two in South Korea, two in Australia, one in Singapore, and one in Hongkong). Only
three B.1.1.214 cases have been detected outside of Japan (two in Australia and one in the
USA). Among our 198 samples, some harboring other lineages of Clade 20B were also detected:
15 from the B.1.1.285 lineage, 10 from the B.1.1.114 lineage, 4 from the B.1.1.119 lineage,
and 1 from the B.1.1.163 lineage.
The B.1.346 lineage belonging to clade 20C that likely emerged in New York10 was detected in one patient with confirmed
COVID-19 infection at a hospital in the Kanto region in November 2020 (Fig. 2, B.1.346). The patient had no history of overseas travel or
known contact with anyone who had travelled abroad. This strain has 18 single nucleotide
substitutions compared with the original Wuhan SARS-CoV-2 sequence (ID. NC_045512.2). The
functional relevance of the single nucleotide substitutions is summarized in Table 1.

Table 1.
Single nucleotide substitutions and their functional relevance in B.1.346 in Clade
20C compared with that of the Wuhan strain
| Position in reference to the Wuhan strain |
Reference allele |
Variant allele |
Protein |
Amino acid change |
Annotation |
Note |
| 241 |
C |
T |
5′UTR |
NA |
NA |
|
| 1059 |
C |
T |
NSP2 |
T85I |
Non-structural protein 2 |
20C signature |
| 3037 |
C |
T |
NSP3 |
F106F |
Predicted phosphoesterase,
papain-like
proteinase |
20A signature |
| 6948 |
A |
C |
NSP3 |
N1410T |
Predicted phosphoesterase,
papain-like
proteinase |
Common among five strains from the western USA |
| 12820 |
A |
G |
NSP9 |
L45L |
ssRNA-binding protein |
|
| 14408 |
C |
T |
NSP12b |
P314L |
RNA-dependent RNA polymerase,
post-ribosomal
frameshift |
20A signature |
| 14587 |
G |
T |
NSP12b |
A374S |
RNA-dependent RNA polymerase,
post-ribosomal
frameshift |
|
| 15324 |
C |
T |
NSP12b |
N619N |
RNA-dependent RNA polymerase,
post-ribosomal
frameshift |
Common among five strains from the western USA |
| 15403 |
T |
C |
NSP12b |
R645R |
RNA-dependent RNA polymerase,
post-ribosomal
frameshift |
Common among five strains from the western USA |
| 16762 |
C |
T |
NSP13 |
L176F |
Helicase |
|
| 17637 |
A |
G |
NSP13 |
K467 K |
Helicase |
|
| 17880 |
A |
G |
NSP13 |
Q548Q |
Helicase |
|
| 20762 |
C |
T |
NSP16 |
T35I |
2’-O-Ribose methyltransferase |
|
| 23403 |
A |
G |
S |
D614G |
Spike |
20A signature |
| 24337 |
C |
T |
S |
N925N |
Spike |
Common among five strains from the western USA |
| 25563 |
G |
T |
ORF3a |
Q57H |
ORF3a protein |
20C signature |
| 26735 |
C |
T |
M |
Y71Y |
Membrane |
Common among four strains from the western USA |
| 28887 |
C |
T |
N |
T205I |
Nucleocapsid protein |
|
NSP, non-structural polyprotein; ORF, open reading frame; ssRNA, single-strand RNA; NA,
not assessed.
In total, 159 strains belonging to the B.1.346 lineage have been deposited in the GISAID
database (see Acknowledgment Table). Most of these strains were found in the western United
States. Detailed examination of the phylogenetic tree revealed that four strains were very
closely related (Fig. 2). The genomic data
strongly suggest that this strain was imported into Japan across the quarantine barrier.
GISAID data from Japan indicate that the last clade 20C strains detected in Japan were in
March and May 2020, but none were detected thereafter, except for quarantine cases. These
findings support the notion that the newly identified clade 20C strain in this study was
indeed imported from overseas.
Comparison of the fingerprinting patterns of the B.1.346 lineage strains detected in Japan
in this study, those detected in the western United States, and a representative clade 20C
strain detected in Japan in the spring of 2020 (Fig.
3) further supports the notion that the strain of B.1.346 lineage belonging to
clade 20C was imported into Japan across the quarantine barrier from the western United
States. There is no evidence that the newly detected clade 20C strain in the Kanto region
shows altered transmissibility or virulence, unlike variants of concern such as 501Y.V1
(B.1.1.7) in the UK and 501Y.V2 (B.1.351) in South Africa.
Discussion
Through lineage analysis of 198 whole viral genomes accumulated from 13 hospitals in the
Kanto region, we have herein concluded the following. First, most of the strains belonged to
B.1.1.284 and B.1.1.214. Second, at least one strain was imported from the USA. We could not
detect the importation route of this foreign strain (B.1.346), but we guess that it likely
slipped through airport quarantine and spread human-to-human.
The fact that only two strains were predominant in the Kanto region supports the notion
that Japan’s quarantine system was relatively successful after the national lockdown in
April and May of 2020. The observation of an extremely limited number of predominant
lineages (i.e., only two) is in sharp contrast to the situation in the UK and other
countries where numerous strains are prevalent.11,12 In view
of the emergence of potentially virulent strains (501Y) in the UK and South Africa, a
further thorough and strict quarantine system is warranted in Japan.
The implications of the detection in Japan of a viral strain prevalent in the western
United States are twofold. First, no quarantine policy is perfect. If the policy is adjusted
to become less strict, close follow-up genomic monitoring will be mandatory. Because of the
internationally unprecedented uniformity of the domestic viral strains in Japan, detection
of foreign strains may be relatively easy, as exemplified by the single patient in the
current report. Second, if foreign strains do enter Japan in the near future, it may be
relatively easy to pinpoint the geographic origin of the incoming strain by the number of
nucleotide substitutions in the SARS-CoV-2 genome.
Overall, the viral genomic monitoring system for in-hospital infections reported herein
enabled us to swiftly unravel regional and national trends. In Japan, a national centralized
network system composed of the National Institute of Infectious Diseases in Tokyo, public
health centers, and public health institutes was put in place for the purpose of viral
genome surveillance. This report illustrates that an agile decentralized network composed of
multiple hospitals mutually sharing molecular genomic data in near real-time provides robust
benefits to public health under the conditions of the present COVID-19 pandemic. Molecular
genomic data obtained through such a system can be swiftly reflected in the national
decision-making process for public health practices, including the strictness of quarantine
measures or the implementation of lockdowns in response to the detection of SARS-CoV-2
variants of concern.
Data sharing.
We downloaded the full nucleotide sequences of the SARS-CoV-2 genomes from the GISAID
database (https://www.gisaid.org/). A table of the contributors is available below
(Acknowledgment Table). We have uploaded the full nucleotide sequences of our cohort to the
GISAID database.
Acknowledgments
We thank all the patients and healthcare workers who have fought against COVID-19. This
work was supported by the Keio Donner Project and is devoted to the late Professor
Shibasaburo Kitasato, the founder of Keio University School of Medicine. We also thank SRL,
Inc. Funding provided by Keio Gijuku Academic Development Funds and the Japan Agency for
Medical Research Development (AMED JP20he0622043, K.K. as the Lead) was used to acquire
consumables and to carry out deep sequencing of viral genomes. The cost of consumables was
also supported in part by Ryoshoku-Kenkyu-kai, which aims to tackle infections disease
control (M.S.). The design and data analyses of this study were performed independently of
the funding agencies.
Conflicts of Interest
The authors have no conflicts of interest to report.
References
- 1. Takenouchi T, Iwasaki YW, Harada S, Ishizu H,
Uwamino Y, Uno S, Osada A, Abe K, Hasegawa N, Murata M, Takebayashi T, Fukunaga K, Saya H,
Kitagawa Y, Amagai M, Siomi H, Kosaki K: Clinical utility of SARS-CoV-2 whole genome
sequencing in deciphering source of infection. J Hosp Infect 2021; 107: 40–44.
DOI:10.1016/j.jhin.2020.10.014
- 2. du Plessis L, McCrone JT, Zarebski AE, Hill V,
Ruis C, Gutierrez B, Raghwani J, Ashworth J, Colquhoun R, Connor TR, Faria NR, Jackson B,
Loman NJ, O’Toole Á, Nicholls SM, Parag KV, Scher E, Vasylyeva TI, Volz EM, Watts A,
Bogoch II, Khan K, Aanensen DM, Kraemer MU, Rambaut A, Pybus OG, COVID-19 Genomics UK
(COG-UK) Consortium: Establishment and lineage dynamics of the SARS-CoV-2 epidemic in the
UK. Science 2021; 371: 708–712. PMID:33419936,
DOI:10.1126/science.abf2946
- 3.Davies N, Barnard R, Jarvis C,et al. Estimated
transmissibility and severity of novel SARS-CoV-2 Variant of Concern 202012/01 in England.
[Preliminary] CMMID repository 23–12-2020 first online.
- 4. European Centre for Disease Prevention and Control:
Risk related to spread of new SARS-CoV-2 variants of concern in the EU/EEA. Stockholm,
2020.
- 5. Sekizuka T, Itokawa K, Hashino M, Kawano-Sugaya
T, Tanaka R, Yatsu K, Ohnishi A, Goto K, Tsukagoshi H, Ehara H, Sadamasu K, Taira M,
Shibata S, Nomoto R, Hiroi S, Toho M, Shimada T, Matsui T, Sunagawa T, Kamiya H, Yahata Y,
Yamagishi T, Suzuki M, Wakita T, Kuroda M: A genome epidemiological study of SARS-CoV-2
introduction into Japan. MSphere 2020; 5: e00786-20. PMID:33177213,
DOI:10.1128/mSphere.00786-20
- 6. Sekizuka T, Itokawa K, Kageyama T, Saito S,
Takayama I, Asanuma H, Nao N, Tanaka R, Hashino M, Takahashi T, Kamiya H, Yamagishi T,
Kakimoto K, Suzuki M, Hasegawa H, Wakita T, Kuroda M: Haplotype networks of SARS-CoV-2
infections in the Diamond Princess cruise ship outbreak. Proc Natl Acad Sci USA 2020; 117:
20198–20201. PMID:32723824, DOI:10.1073/pnas.2006824117
- 7. Abe K, Kabe Y, Uchiyama S,et al: Severity of
COVID-19 is inversely correlated with increased number counts of non-synonymous mutations
in Tokyo. medRxiv 2020; https://doi.org/.DOI:10.1101/2020.11.24.20235952
- 8.Hadfield J, Megill C, Bell SM, Huddleston J,
Potter B, Callender C, Sagulenko P, Bedford T, Neher RA: Nextstrain: real-time tracking of
pathogen evolution. Bioinformatics 2018; 34: 4121-4123.
- 9. Rambaut A, Holmes EC, O’Toole Á, Hill V, McCrone
JT, Ruis C, du Plessis L, Pybus OG: A dynamic nomenclature proposal for SARS-CoV-2
lineages to assist genomic epidemiology. Nat Microbiol 2020; 5: 1403–1407. PMID:32669681,
DOI:10.1038/s41564-020-0770-5
- 10. Zhang W, Davis B, Chen S,et al: Emergence of a
novel SARS-CoV-2 strain in Southern California, USA. medRxiv 2021;
doi.org/.DOI:10.1101/2021.01.18.21249786
- 11. Popa A, Genger JW, Nicholson MD, Penz T, Schmid
D, Aberle SW, Agerer B, Lercher A, Endler L, Colaço H, Smyth M, Schuster M, Grau ML,
Martínez-Jiménez F, Pich O, Borena W, Pawelka E, Keszei Z, Senekowitsch M, Laine J, Aberle
JH, Redlberger-Fritz M, Karolyi M, Zoufaly A, Maritschnik S, Borkovec M, Hufnagl P, Nairz
M, Weiss G, Wolfinger MT, von Laer D, Superti-Furga G, Lopez-Bigas N, Puchhammer-Stöckl E,
Allerberger F, Michor F, Bock C, Bergthaler A: Genomic epidemiology of superspreading
events in Austria reveals mutational dynamics and transmission properties of SARS-CoV-2.
Sci Transl Med 2020; 12: eabe2555. PMID:33229462,
DOI:10.1126/scitranslmed.abe2555
- 12. Lemieux JE, Siddle KJ, Shaw BM, Loreth C,
Schaffner SF, Gladden-Young A, Adams G, Fink T, Tomkins-Tinch CH, Krasilnikova LA, DeRuff
KC, Rudy M, Bauer MR, Lagerborg KA, Normandin E, Chapman SB, Reilly SK, Anahtar MN, Lin
AE, Carter A, Myhrvold C, Kemball ME, Chaluvadi S, Cusick C, Flowers K, Neumann A, Cerrato
F, Farhat M, Slater D, Harris JB, Branda JA, Hooper D, Gaeta JM, Baggett TP, O’Connell J,
Gnirke A, Lieberman TD, Philippakis A, Burns M, Brown CM, Luban J, Ryan ET, Turbett SE,
LaRocque RC, Hanage WP, Gallagher GR, Madoff LC, Smole S, Pierce VM, Rosenberg E, Sabeti
PC, Park DJ, MacInnis BL: Phylogenetic analysis of SARS-CoV-2 in Boston highlights the
impact of superspreading events. Science 2021; 371: eabe3261.
PMID:33303686