Abstract
Inflammation and immune responses after tissue injury play pivotal roles in the
resolution of inflammation, tissue recovery, fibrosis, and remodeling. Regulatory T cells
(Tregs) are responsible for immune tolerance and are usually activated in secondary
lymphatic tissues. Activated Tregs subsequently regulate effector T cell and dendritic
cell activation. For clinical applications such as the suppression of both autoimmune
diseases and the rejection of transplanted organs, methods to generate stabilized
antigen-specific Tregs are required. For this purpose, transcriptional and epigenetic
regulation of Foxp3 expression has been investigated. In addition to conventional Tregs,
there are some Tregs that reside in tissues and are called tissue Tregs. Tissue Tregs
exhibit tissue-specific functions that contribute to the maintenance of tissue homeostasis
and repair. Such tissue Tregs could also be useful for Treg-based cell therapy. We
recently discovered brain Tregs that accumulate in the brain during the chronic phase of
ischemic brain injury. Brain Tregs resemble other tissue Tregs, but are unique in
expressing neural cell-specific genes such as the serotonin receptor (Htr7); consequently,
brain Tregs respond to serotonin. Here, we describe our experiences in the use of Tregs to
suppress graft-versus-host disease and to promote neural recovery after stroke.
Introduction
Regulatory T cells (Tregs) suppress unwanted immunity against a variety of antigens,
including self-antigens, commensal bacteria-derived antigens, and environmental
allergens,1 thereby preventing the
development of autoimmune diseases, colitis, and allergies.2,3,4 Tregs
express Forkhead box P3 (Foxp3) as a major master transcription factor and suppress
excessive immune responses by recognizing various self-antigens and foreign
antigens.5 Tregs are generated mainly
via two different pathways. The first is direct development from CD4 and CD8 double-positive
T cells in the thymus. Tregs that develop via this route are called thymus-derived Tregs
(tTregs), or naturally occurring Tregs (nTregs). tTregs are believed to develop from
precursor thymic T cells by recognizing self-antigen-MHC complexes expressed on thymic
antigen-presenting cells with relatively high avidity. Therefore, tTregs express a T cell
receptor (TCR) repertoire with a bias for self, and are important for the prevention of
autoimmunity.6,7 The second pathway of Treg generation is
differentiation from naïve CD4+ T cells in the periphery upon antigen stimulation
with an appropriate combination of cytokines, including interleukin-2 (IL-2) and
transforming growth factorTGF-β).8,9 Tregs
produced in this way are called induced Tregs (iTregs) or peripherally induced Tregs
(pTregs); the term “iTreg” is often used for Tregs generated in vitro,
whereas the term “pTreg” indicates Tregs generated from naïve T cells in
vivo. It is estimated that tTregs compose most of the systemic Treg population,
whereas pTregs are highly enriched in certain organs, including the gut and maternal
placenta.8,10 TGF-β is essential for the generation of both
iTregs and pTregs,11 and lamina propria
CD103-positive dendritic cells (LPDCs) are major producers of TGF-β in the
intestine.12,13 Furthermore, several commensal bacteria
reportedly stimulate CD103+ LPDCs and increase TGF-β production.12
Various mechanisms of immune suppression by Tregs have been proposed: IL-2 consumption by
CD25 expression, suppression of co-stimulation by CTLA4 expression, and suppression of
inflammation by anti-inflammatory cytokine IL-10 and TGF-β.11,14 In recent years, in addition to its immune suppression functions, Tregs
localized in non-lymphoid tissues have attracted attention in various fields. Tregs are
either present in various tissues at steady state or accumulate after tissue injury; they
play important roles in tissue homeostasis and repair by interacting with tissue cells.
These Tregs are called tissue Tregs; they exhibit common properties among tissues, but they
also have characteristics specialized for each tissue.15,16 In this
review article, we describe the molecular mechanisms that govern Treg differentiation and
maintenance, focusing on the roles of transcription factors and epigenetic modifications. We
will describe an example application of Tregs in a murine graft-versus-host disease (GVHD)
model.17 We will also review tissue
Tregs and their application to treat diseases of the central nervous system.18,19
Mechanism of Treg Development
Tregs are characterized by the expression of transcription factor Foxp3, which plays
crucial roles in the differentiation, maintenance, and function of Tregs. The importance of
Foxp3 is evident, because differentiation of tTregs and pTregs is severely impaired in
humans and mice with Foxp3 loss-of-function mutations, leading to death as
a result of severe inflammatory diseases.2,3,4 Given the
critical roles of Foxp3 in Treg biology, the molecular mechanisms underlying the induction
of this transcription factor have been extensively analyzed (Fig. 1).20

In addition to the promoter, previous studies identified several intronic enhancers at the
Foxp3 gene locus that are important for Treg differentiation.21 Each enhancer was shown to differentially
contribute to tTreg and pTreg differentiation. The enhancers were designated as conserved
noncoding sequence (CNS) 0, 1, 2, and 3, with the numbers reflecting their distances from
the transcriptional start site (Fig. 1).22,23 CNS0 was identified as a Satb1-binding site.23 Satb1 is a global genome organizer that
induces both transcriptional regulation and epigenetic regulation via the formation of novel
nuclear architectures.24 Satb1 is believed
to function as a pioneering element required for the subsequent activities of the other CNS
elements that lead to the initiation of Foxp3 expression.23
The CNS1 enhancer contains binding sites for transcription factors, including Smads, NFAT,
AP-1, and retinoic acid receptor (RAR) (Fig.
1).20 Retinoic acid (RA)
contributes to high and stable Foxp3 transcription through RAR.25 CD103+ dendritic cells (DCs) in the
lamina propria and mesenteric lymph nodes express the enzyme retinal aldehyde dehydrogenase,
which synthesizes RA.26 TGF-β induces
binding of Smad2/3 to CNS1, which is a critical step for iTreg/pTreg
differentiation.7,23 Smad2 and Smad3 are redundantly essential for
the induction of Foxp3 by TGF-β stimulation.9 TGF-β1-deficient mice and Smad2/3 double-deficient mice exhibited
relatively normal tTreg development in the thymus, but showed significantly reduced numbers
of pTregs.9,27 These reports confirmed the essential role of
TGF-β–Smad2/3 signaling in pTreg development. Accelerated mucosal T helper type 2 (Th2)
cell-type inflammation and abortion were observed in CNS1-deleted mice, suggesting the
importance of pTregs for suppression of excessive immunity against commensal bacteria and
fetuses.28,29
The CNS2 enhancer contains binding sites for transcription factors, including Stat5, NFAT,
Runx1/Cbfβ, CREB, and Foxp3 (Fig. 2).30 For example, IL-2 signaling is critical for
tTreg and pTreg differentiation, and Stat5-responsive elements exist in the
Foxp3 CNS2 and promoter region.31,32,33,34,35,36 CNS2
enhancer is important for the maintenance of Foxp3 expression, particularly
under inflammatory conditions in which Tregs are exposed to inflammatory cytokines and
stronger TCR stimulation.37,38 The
CNS2 locus is highly enriched with CpG sites, and the methylation status is an especially
important determinant of the activity of this enhancer. In tTregs, the CpG sites are fully
demethylated, which contributes to sustained expression of Foxp3 in tTregs and the stability
of the tTreg lineage phenotypes (Fig.
2).37,38,39,40,41,42,43 DNA
demethylation at this locus is also observed in pTregs, albeit with slightly reduced
penetrance compared with tTregs,39,44,45 rendering
pTregs a stable subset.39,45
However, this region is rarely demethylated in in vitro-generated iTreg
cells and, consequently, iTregs are highly unstable.39,40 Full
methylation of this locus was also reported to prevent abnormal Foxp3 induction in
non-Tregs, including CD8+ T cells and natural killer cells.41,46,47

The CNS3 enhancer reportedly contains binding sites for c-Rel and is important for the
differentiation of tTregs and pTregs (Fig.
1).21,48 In CNS3-enhancer-deleted mice, Treg
development was severely impaired, and c-Rel was critical for tTreg development at the
Foxp3 promoter.49 We
found that TFAR6, an adaptor protein that activates NF-κB (including c-Rel), is essential
for stable Foxp3 expression.50
Recruitment of Nr4a Factors to the Promoter of Foxp3
The promoter at the Foxp3 locus is activated by various types of stimuli,
including TCR stimulation and cytokine signaling. We recently found that members of the Nr4a
nuclear receptor transcription factor family of nuclear orphan receptors have essential
roles in Treg differentiation by directly acting on the Foxp3
promoter51,52,53 (Fig. 1). The Nr4a
family is composed of the closely related molecules Nr4a1, Nr4a2, and Nr4a3, which belong to
a nuclear receptor superfamily.20 The
expression of Nr4a factors is highly enriched in tTregs compared with other T-cell
subsets.54,55,56,57 Nr4a factors act directly on the Foxp3 promoter and
strongly induce Foxp3 expression.52 Nr4a
family members are redundantly important for tTreg development in the thymus, as revealed by
the nearly complete loss of Tregs in mice with the specific deletion of all three Nr4a
factors in T cells.51 Nr4a factors are
also necessary for in vitro iTreg differentiation. Expression of all Nr4a
factors is induced by TCR stimulation, suggesting that Nr4a factors are crucial mediators of
TCR signaling in Foxp3 induction.56,58,59 Forced
activation of Nr4a in T cells is sufficient for Foxp3 expression without strong TCR
stimulation.51 The whole promoter
region also regulates Foxp3 expression by sensing both cytokine and TCR signaling, which is
mediated by transcription factors including Nr4a factors.
After tTreg development, Nr4a factors are expressed at high levels in the periphery. A
knockout mouse strain in which all Nr4a genes were specifically deleted in Tregs showed a
global reduction of genes exclusively expressed in Tregs, including Foxp3,
Il2r, and Ikzf4. These mice also developed Th2-type
systemic autoimmune diseases. Nr4a-deficient Tregs easily lost Foxp3 expression and became
Th2 and T follicular helper cells. These findings demonstrate that Nr4a controls a novel
genetic program required for Treg cell maintenance and function.60
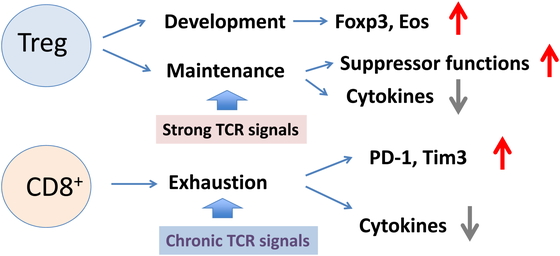
In addition to Treg development, Nr4a factors were recently reported to be important for
CD8+ T cell exhaustion61,62,63. “T cell
exhaustion” is a phenomenon of dysfunction or physical elimination of antigen-specific T
cells reported in chronic viral infections as well as in cancer. Unlike conventional T
cells, exhausted T cells lose the ability to proliferate, to produce cytokines, and effector
functions in response to antigens. Exhausted T cells express high levels of NR4a
factors.61 The phenotypes of T cell
exhaustion partly overlap with anergic phenotypes of Tregs, including a high expression of
checkpoint receptors (e.g., PD-1, Tim-3, and CTLA4) and low expression of cytokines such as
IL-2 and IFNγ. Inhibitors of Nr4a could potentiate anti-tumor immunity by reducing the
number of Tregs and enhancing CD8+ T cell activity.64 Therefore, NR4a factors seem to be important for immune
tolerance by conferring anergic phenotypes to both CD4+ and CD8+ T
cells (Fig. 3).
Treg-specific Epigenetic Modifications and Stability of Tregs
Ohkura et al. showed that the establishment of a Treg-specific CpG hypomethylation pattern
[called Treg-specific demethylated regions (TSDRs)] is crucial for Treg
development.41 TSDRs are distributed
in genes that are important for Treg differentiation and function, including the CNS2
enhancer (Fig. 1) of Foxp3 and
specific regions in Ctla4, Il2ra (encoding CD25),
Ikzf4 (encoding Eos), and Tnfrs18 (encoding GITR).
Demethylation of the upstream enhancer, which is close to CNS0, was also observed in
tTregs.23,65,66 Without the establishment of a TSDR hypomethylation pattern,
even Foxp3-positive cells cannot acquire full suppressive activity. Instead, Foxp3-positive
TSDR-methylated cells, which are similar to in vitro-generated iTregs, show
highly unstable Treg-associated gene expression. Demethylation of CNS2 at the
Foxp3 locus is believed to be maintained by stable binding of Foxp3 and
the Cbf-β-Runx1 complex or CREB/ATF to demethylated CNS2.
Recent reports revealed that members of the ten-eleven translocation (Tet) family of
demethylation factors have important roles in CpG demethylation at CNS2 in Tregs.67,68,69 Mice in which both Tet1 and Tet2 were specifically deleted in T cells or
Tregs developed autoimmune diseases as a result of reduced Treg differentiation and
function.70 Moreover, Stat5 interacts
with Tet1 and Tet2 and recruits them to CNS2, as well as to the promoter and CNS1, thereby
mediating CpG demethylation at those loci.70 In Tet2/Tet3 double-deficient mice, Tet2 and Tet3
proteins were shown to mediate demethylation of CNS1 and CNS2 in the Foxp3
locus and other TSDRs in tTregs.66,71,72,73, Hydrogen sulfide promoted the expression of Tet1 and Tet2,
which were recruited to the Foxp3 locus by TGF-β and IL-2 signaling to
maintain Foxp3 demethylation and Treg-associated immune
homeostasis.70 We found that reduced
oxygen concentrations during iTreg induction resulted in the upregulation of Tet enzyme
expression and the promotion of CNS2 demethylation.74
Recent studies have demonstrated that demethylation of CNS2 by Tet enzymes is promoted by
vitamin C in iTregs, leading to stabilized Foxp3 expression.17,65,72,74 Vitamin C
was shown to potentiate Tet activity,75,76 thereby
facilitating demethylation of the Foxp3 CNS2 region and increasing the
stability of Foxp3 expression in TGF-β-induced iTregs.77 Compared with untreated iTregs, iTregs generated under low oxygen
conditions in the presence of vitamin C retained more stable Foxp3 expression in
vitro and in vivo and exhibited stronger suppression activity in
a colitis model.
Use of iTregs to Prevent GVHD
Preparation of iTregs could be an effective strategy for Treg-mediated adoptive
immunotherapy (Fig. 4).74 Antigen-specific Tregs are expected to induce antigen-specific
tolerance, rather than broad immunosuppression. Because iTregs can be efficiently expanded
from naïve T cells, antigen-specific iTregs are a realistic choice for adaptive immune
therapy of autoimmune diseases, suppression of rejection during organ transplantation, and
prevention of GVHD in bone marrow transplantation. We showed that antigen-specific,
polyclonally expanded iTregs induced stronger tolerance than polyclonal tTregs in a heart
transplantation model.78 However, iTregs
generated by conventional methods did not effectively suppress GVHD in an animal model,
because these iTregs were very unstable after adoptive transfer, probably as a result of the
extreme inflammatory conditions. Consequently, we sought an optimal method to generate
stable iTregs that can prevent GVHD in a murine model. Alloantigen-specific iTregs were
generated by coculture of naïve T cells with allogenic DCs in the presence of TGF-β and RA.
By examining various agents and genes, we found that a high concentration of vitamin C
(100 µg/ml) stabilized Foxp3 expression most effectively in adoptively transferred iTregs
under the GVHD environment (Fig. 4A). Vitamin
C-treated iTregs suppressed GVHD symptoms more efficiently than untreated iTregs. Vitamin C
treatment caused almost complete CNS2 DNA demethylation in alloantigen-specific iTregs; such
treatment also reduced iTreg conversion into pathogenic Foxp3-negative cells (Fig. 4B).
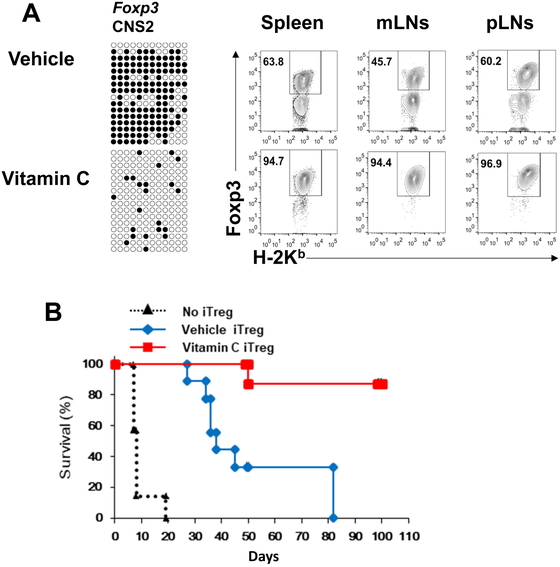
We observed stable, very high FOXP3 expression in human iTregs that underwent vitamin
C-treatment.17 Vitamin C treatment of
iTregs also shows promise for innovative clinical applications of adoptive Treg
immunotherapy. Further understanding of the mechanisms that facilitate the establishment of
a Treg-specific transcriptional program will allow the production of sufficient numbers of
antigen-specific Tregs with appropriate stability.
Tregs Reside in Tissues, Including the Brain
Tregs usually represent about 10% of CD4+ T cells and are considered to exist in
lymphoid tissues and sites of inflammation; however, it has recently been discovered that
they accumulate in various tissues in addition to lymphoid tissues. Tissue Tregs recognize
the self-antigen characteristic of the tissues and have a limited TCR repertoire. Such
tissue-resident Tregs (e.g., in fat, muscle, skin, lung, and intestines) exhibit phenotypes
quite different from those present in lymphoid tissue15,79,80 (Table 1). The following features are common to
various tissue Tregs: high expression of genes such as Il10,
Il1rl1 (encoding ST2, IL-33 receptor), Areg
(amphiregulin), Klrg1, Ctla4, Tigit,
Gata3, Batf, and Irf4 and low
expression of Lef1, Tcf7, and Bcl2
compared with lymphatic tissue Tregs.79,81 Analysis
of the A384T mutation of the Foxp3 gene has shown that BATF is an important regulator of
tissue Tregs.82 However, tissue Tregs also
have tissue-specific features that are induced by the localized microenvironment, and these
features seem to play pivotal roles in the phenotypes of tissue Tregs. The features of
various tissue Tregs are shown in Table
1.
Table 1.
Features of various tissue Tregs
| Tissue |
Transcription factor |
Function |
Specific molecules |
Localization receptor |
References |
| Fat (VAT) |
PPARγ |
Suppression of fat inflammation, metabolic
modifications, aging |
IL-10 |
? |
123,124,125,126 |
| Muscle |
PPARγ |
Proliferation of satellite cells |
Areg |
CCR2 |
127,128,129,130 |
| Heart |
|
Suppression of activation, M2 induction |
IL-10 |
CCR5 |
131,132,133,134 |
| Lung |
|
Suppression of pulmonary fibrosis
Proliferation of alveolar type II cells |
Areg |
CD103 |
135,136,137 |
| Skin |
GATA3 |
Regeneration of hair follicles, skin homeostasis,
wound healing |
Jag1-Notch |
CCR4,6,8, CD103 |
138,139,140,141 |
| Colon |
Helios-RORgt+ |
Suppression of inflammation |
IL-10, CTLA4 |
? |
142 |
|
Helios+Gata3+ |
Tissue repair |
Areg |
? |
143 |
| Brain |
PPARγ |
Suppression of astrogliosis |
Areg, CCN3 |
CCR6,8 |
18,87 |
VAT, visceral adipose tissue.
The number of Tregs in the brain is extremely low under normal conditions. However, the
involvement of Tregs in neuroinflammatory diseases such as multiple sclerosis (MS) has been
intensively studied.83,84 Tregs
from MS patients have defects in their suppression ability owing to a decrease in the
expression levels of CTLA-4, Foxp3, and some genes important for Treg function, and these
defects may contribute to the onset of disease.85 In a mouse model of experimental autoimmune encephalomyelitis, which
mimics MS, symptoms worsen as a result of the depletion or deficiency of Tregs.83 In mice lacking T cells, remyelination is
delayed after lysolecithin injection, suggesting that T cells are required for remyelination
of the central nervous system.86,87 It has
been reported that the secretory protein CCN3 produced by Tregs promotes the differentiation
and myelination of oligodendrocyte precursor cells.87 Previously, Tregs infiltrated into the brain were reported to suppress
neuroinflammation and reduce the severity of experimental stroke at the acute
phase.88 However, the role of Tregs in
stroke has become controversial,89,90 mostly
because the number of Tregs during the acute phase of ischemic brain injury is extremely low
(less than 100 cells per brain), and antigen-specific proliferation and activation of Tregs
may not occur in such a short period (i.e., within 3 days). Therefore, only a bystander
effect, such as the secretion of IL-10, may be observed during the acute phase of ischemic
stroke.
Tregs in Ischemic Brain Injury
We have been studying the immune responses after ischemic brain injury. The resulting brain
damage causes neuronal cell death and destruction of neuronal circuits, resulting in
impaired movement, sensation, and higher brain functions. Although thrombolytic therapy
delivered as intravenous recombinant tissue-plasminogen activator alleviates ischemic brain
damage, to be effective, it should be administered within 4.5 h after the onset. After this
very early period, only rehabilitation is available as the main treatment for promoting
functional recovery for most ischemic stroke patients.
Sterile inflammation is usually associated with tissue injury and cell death. In the case
of ischemic stroke, inflammation occurs both in humans and in mouse models, and it is
suggested that neuroinflammation is an attractive treatment target for reducing brain
damage.91,92,93,94 Currently, innate immune cells, including microglia, macrophages, and γδ
T cells, are thought to play major roles in neuroinflammation after stroke, because
inflammation is apparent within a few days after stroke onset95,96 (Fig. 5). By day 3 after
disease onset, IL-17 secreted by γδ T cells contributes to damage in the ischemic penumbra
region.95,97 Then, after 3–4 days, infiltrated M1-type
macrophages are converted to M2-type repairing macrophages, and few symptoms of inflammation
are observed 1 week after stroke98 This is
mostly because M2-type macrophages facilitate the scavenging of necrotic cells and tissue
debris and support neural repair by producing trophic cytokines such as IGF-I (Fig. 5). However, our group and others have identified
a massive accumulation of lymphoid cells, including Tregs, in the chronic phase, more than 2
weeks after stroke onset.99,100 Tregs
account for approximately half of CD4+ T cells localized inside and around the
cerebral infarction lesion in close proximity to scar-forming astrocytes and surviving
neuronal cells (Fig. 5).

Characterization of Brain Tregs
Like other tissue Tregs, brain Tregs are Helios+ KLRG1+ tTregs; they
possess a unique TCR repertoire and express high levels of CTLA-4, PD-1, Areg, and ST2.
Moreover, similar to other tissue Tregs, TCR signaling, IL-2, and IL-33 are essential for
the proliferation of brain Tregs (Fig. 6). In the
brain, both astrocytes and oligodendrocytes express IL-33. It is notable that IL-33
reportedly promotes tissue recovery after central nervous system injury87 and induces M2 type macrophage-related
genes.101 It is also highly possible
that brain Tregs are involved in the repair function of IL-33 in the central nervous system
(Fig. 6).

Moreover, brain Tregs express several unique genes, such as central nervous system-related
genes, that are not present in other tissue Tregs. In particular, brain Tregs express
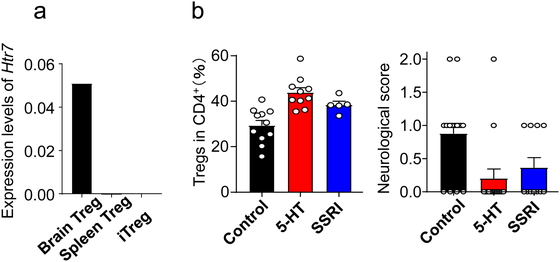
serotonin receptor 7 (Htr 7), which raises cAMP levels (Fig. 6).102 cAMP promotes the
proliferation of Tregs and potentiates their functionality.103 The administration of serotonin or a selective serotonin
reuptake inhibitor (SSRI) during the chronic phase after stroke increases the number of
brain Tregs and improves neurological symptoms (Fig.
7). Compared with wild type Tregs, Htr7-deficient Tregs do not
increase in number in the brain and cannot reduce neurological deficits after transfer into
T cell-deficient mice. As expected, in vitro, Tregs isolated from the
ischemic brain proliferate and are activated in an Htr7-dependent manner. Brain Tregs also
express CCR6 and CCR8 and infiltrate the brain with the expression of CCL20 and CCL1 in the
cerebral infarct area. Intraventricular injections of CCL1 and CCL20 increase the number of
Tregs, leading to improved neurological recovery.
The depletion or reduction of brain Tregs causes excessive activation of astrocytes and
apoptosis of neurons in the motor cortex. Therefore, brain Tregs reduce the activation of
astrocytes. Excess activation of astrocytes, so-called astrogliosis, is thought to lead to a
delay in the recovery of motor function after brain or spinal cord injury.104 Although astrogliosis is necessary for
forming scar tissue to protect neuronal cells from necrotic areas, excessive activation of
astrocytes increases neurotoxic factors, resulting in neural cell damage and inhibition of
neuronal outgrowth.105
Inflammatory cytokines such as IL-6 are important for the activation of
astrocytes,106 and brain Tregs
suppress IL-6 levels in vivo and in vitro. Areg is known
to suppress the production of inflammatory cytokines, including IL-6 and TNFα, in several
inflammatory diseases.107 We found that
Areg suppresses IL-6 production from microglia and astrocytes in vivo
(Fig. 6). Therefore, Areg is an important
functional molecule of brain Tregs. Areg may also be directly involved in the proliferation
of neural stem cells.108
Clinical Implications of Tregs for Central Nervous System Diseases
It has not yet been demonstrated that Tregs accumulate in the brains of ischemic stroke
patients. However, in human cerebral infarctions, correlations between the number of
peripheral Tregs, the Treg/Th17 ratio, or IL-17/IL-10 levels and the severity of stroke have
been reported.109,110,111,112 It is notable that stroke patients show greater immunoreactivity to
brain self-antigens.113 SSRI is known to
ameliorate neurological symptoms after stroke onset.114,115
Therefore, it is highly likely that brain Tregs work on neural repair in human stroke
patients. Consistently, a pilot study of FTY720 (fingolimod), an immuno-modulating drug, in
stroke patients revealed that the administration of FTY720 was only effective within 3 days
after stroke onset.116
Neural inflammation occurs not only in cerebral infarction but also in various types of
damage to cerebrospinal tissues, such as spinal cord injury, autoimmune diseases such as
multiple sclerosis, and in neurodegenerative diseases such as Alzheimer’s disease and
Parkinson’s disease.117,118,119 It is
possible that these various types of neural inflammation also trigger acquired immunity and
that brain Tregs infiltrate and accumulate in the central nervous system, thereby playing an
important role in relieving neurological symptoms.120 Tregs in such central nervous system diseases have not been well
characterized; however, the Tregs of MS patients were found to proliferate as a result of
serotonin stimulation.121 Therefore,
Tregs present in the central nervous system may be similar to the brain Tregs that we have
characterized.
Conclusion
In summary, our knowledge of Treg development and stability has been expanded by recent
research. We have established methods for the generation of stable antigen-specific Tregs to
suppress GVHD in mice. We also showed that the number of brain Tregs was increased by
chemokines or serotonin and promoted neuronal recovery after ischemic brain injury. The
molecular mechanisms whereby Tregs acquire brain-specific characteristics, including
serotonin receptor expression, remain to be clarified. The identification of self-antigens
and the induction of brain-specific Tregs may also be useful in therapies for relieving
symptoms not only in cerebral infarction but also in other central nervous system diseases.
In the future, it may be possible to administer autologous Tregs directly into the brain for
the treatment of cerebral inflammation.122 More than 25 years have passed since Tregs were discovered. Now that
elucidation of the mechanisms of Treg differentiation, maintenance, and function are almost
complete, the era of therapeutic application has begun.
Acknowledgments
This work was supported by JSPS KAKENHI (S) JP17H06175, Challenging Research (P)
JP18H05376, and AMED-CREST JP18gm0510019 and JP19gm1110009 grants to AY; JSPS KAKENHI
17K15667, 19H04817, and 19K16618 to MI; the Takeda Science Foundation; the Uehara Memorial
Foundation; the Kanae Foundation; and the SENSHIN Medical Research Foundation, Keio Gijuku
Academic Development Funds.
Conflict of Interest
The authors declare no competing financial interests.
References
- 1. Sakaguchi S, Yamaguchi T, Nomura T, Ono M:
Regulatory T cells and immune tolerance. Cell 2008; 133: 775–787. PMID:18510923,
DOI:10.1016/j.cell.2008.05.009
- 2. Bennett CL, Christie J, Ramsdell F, Brunkow ME,
Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD: The immune
dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by
mutations of FOXP3. Nat Genet 2001; 27: 20–21. PMID:11137993,
DOI:10.1038/83713
- 3. Chatila TA, Blaeser F, Ho N, Lederman HM,
Voulgaropoulos C, Helms C, Bowcock AM: JM2, encoding a fork head-related protein, is
mutated in X-linked autoimmunity–allergic disregulation syndrome. J Clin Invest 2000; 106:
R75–R81. PMID:11120765, DOI:10.1172/JCI11679
- 4. Wildin RS, Ramsdell F, Peake J, Faravelli F,
Casanova JL, Buist N, Levy-Lahad E, Mazzella M, Goulet O, Perroni L, Dagna Bricarelli F,
Byrne G, McEuen M, Proll S, Appleby M, Brunkow ME: X-linked neonatal diabetes mellitus,
enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet
2001; 27: 18–20. PMID:11137992, DOI:10.1038/83707
- 5. Hori S, Nomura T, Sakaguchi S: Control of
regulatory T cell development by the transcription factor Foxp3. Science 2003; 299:
1057–1061. PMID:12522256, DOI:10.1126/science.1079490
- 6. Jordan MS, Boesteanu A, Reed AJ, Petrone AL,
Holenbeck AE, Lerman MA, Naji A, Caton AJ: Thymic selection of CD4+CD25+ regulatory T
cells induced by an agonist self-peptide. Nat Immunol 2001; 2: 301–306. PMID:11276200,
DOI:10.1038/86302
- 7. Apostolou I, Sarukhan A, Klein L, von Boehmer H:
Origin of regulatory T cells with known specificity for antigen. Nat Immunol 2002; 3:
756–763. PMID:12089509, DOI:10.1038/ni816
- 8. Kanamori M, Nakatsukasa H, Okada M, Lu Q,
Yoshimura A: Induced regulatory T cells: their development, stability, and applications.
Trends Immunol 2016; 37: 803–811. PMID:27623114,
DOI:10.1016/j.it.2016.08.012
- 9. Takimoto T, Wakabayashi Y, Sekiya T, Inoue N,
Morita R, Ichiyama K, Takahashi R, Asakawa M, Muto G, Mori T, Hasegawa E, Shizuya S, Hara
T, Nomura M, Yoshimura A: Smad2 and Smad3 are redundantly essential for the
TGF-beta-mediated regulation of regulatory T plasticity and Th1 development. J Immunol
2010; 185: 842–855. PMID:20548029, DOI:10.4049/jimmunol.0904100
- 10. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara
T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, Taniguchi T, Takeda K, Hori S, Ivanov
II, Umesaki Y, Itoh K, Honda K: Induction of colonic regulatory T cells by indigenous
Clostridium species. Science 2011; 331: 337–341. PMID:21205640,
DOI:10.1126/science.1198469
- 11. Yoshimura A, Wakabayashi Y, Mori T: Cellular and
molecular basis for the regulation of inflammation by TGF-β. J Biochem 2010; 147: 781–792.
PMID:20410014, DOI:10.1093/jb/mvq043
- 12. Kashiwagi I, Morita R, Schichita T, Komai K,
Saeki K, Matsumoto M, Takeda K, Nomura M, Hayashi A, Kanai T, Yoshimura A: Smad2 and Smad3
inversely regulate TGF-β autoinduction in Clostridium butyricum-activated dendritic cells.
Immunity 2015; 43: 65–79. PMID:26141582, DOI:10.1016/j.immuni.2015.06.010
- 13. Kurebayashi Y, Baba Y, Minowa A, Nadya NA, Azuma
M, Yoshimura A, Koyasu S, Nagai S: TGF-β-induced phosphorylation of Akt and Foxo
transcription factors negatively regulates induced regulatory T cell differentiation.
Biochem Biophys Res Commun 2016; 480: 114–119. PMID:27697523,
DOI:10.1016/j.bbrc.2016.09.153
- 14. Wing JB, Sakaguchi S: Multiple Treg suppressive
modules and their adaptability. Front Immunol 2012; 3: 178. PMID:22754556,
DOI:10.3389/fimmu.2012.00178
- 15. Panduro M, Benoist C, Mathis D: Tissue Tregs.
Annu Rev Immunol 2016; 34: 609–633. PMID:27168246,
DOI:10.1146/annurev-immunol-032712-095948
- 16. Hong J, Kim BS: Regulatory T cell-mediated tissue
repair. Adv Exp Med Biol 2018; 1064: 221–233. PMID:30471036,
DOI:10.1007/978-981-13-0445-3_14
- 17. Kasahara H, Kondo T, Nakatsukasa H, Chikuma S,
Ito M, Ando M, Kurebayashi Y, Sekiya T, Yamada T, Okamoto S, Yoshimura A: Generation of
allo-antigen-specific induced Treg stabilized by vitamin C treatment and its application
for prevention of acute graft versus host disease model. Int Immunol 2017; 29: 457–469.
PMID:29126272, DOI:10.1093/intimm/dxx060
- 18. Ito M, Komai K, Mise-Omata S, Iizuka-Koga M,
Noguchi Y, Kondo T, Sakai R, Matsuo K, Nakayama T, Yoshie O, Nakatsukasa H, Chikuma S,
Shichita T, Yoshimura A: Brain regulatory T cells suppress astrogliosis and potentiate
neurological recovery. Nature 2019; 565: 246–250. PMID:30602786,
DOI:10.1038/s41586-018-0824-5
- 19. Ito M, Komai K, Nakamura T, Srirat T, Yoshimura
A: Tissue regulatory T cells and neural repair. Int Immunol 2019; 31: 361–369.
PMID:30893423, DOI:10.1093/intimm/dxz031
- 20. Sekiya T, Nakatsukasa H, Lu Q, Yoshimura A: Roles
of transcription factors and epigenetic modifications in differentiation and maintenance
of regulatory T cells. Microbes Infect 2016; 18: 378–386. PMID:26970203,
DOI:10.1016/j.micinf.2016.02.004
- 21. Zheng Y, Josefowicz S, Chaudhry A, Peng XP,
Forbush K, Rudensky AY: Role of conserved non-coding DNA elements in the Foxp3 gene in
regulatory T-cell fate. Nature 2010; 463: 808–812. PMID:20072126,
DOI:10.1038/nature08750
- 22. Beyer M, Huehn J: Epigenetic orchestration of
thymic Treg cell development. Nat Immunol 2017; 18: 144–146. PMID:28102221,
DOI:10.1038/ni.3660
- 23. Kitagawa Y, Ohkura N, Kidani Y, Vandenbon A,
Hirota K, Kawakami R, Yasuda K, Motooka D, Nakamura S, Kondo M, Taniuchi I,
Kohwi-Shigematsu T, Sakaguchi S: Guidance of regulatory T cell development by
Satb1-dependent super-enhancer establishment. Nat Immunol 2017; 18: 173–183.
PMID:27992401, DOI:10.1038/ni.3646
- 24. Cai S, Han HJ, Kohwi-Shigematsu T:
Tissue-specific nuclear architecture and gene expression regulated by SATB1. Nat Genet
2003; 34: 42–51. PMID:12692553, DOI:10.1038/ng1146
- 25. Mucida D, Park Y, Kim G, Turovskaya O, Scott I,
Kronenberg M, Cheroutre H: Reciprocal TH17 and regulatory T cell differentiation mediated
by retinoic acid. Science 2007; 317: 256–260. PMID:17569825,
DOI:10.1126/science.1145697
- 26. Coombes JL, Siddiqui KR, Arancibia-Cárcamo CV,
Hall J, Sun CM, Belkaid Y, Powrie F: A functionally specialized population of mucosal
CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β- and retinoic acid-dependent
mechanism. J Exp Med 2007; 204: 1757–1764. PMID:17620361,
DOI:10.1084/jem.20070590
- 27. Marie JC, Letterio JJ, Gavin M, Rudensky AY:
TGF-β1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells.
J Exp Med 2005; 201: 1061–1067. PMID:15809351, DOI:10.1084/jem.20042276
- 28. Samstein RM, Josefowicz SZ, Arvey A, Treuting PM,
Rudensky AY: Extrathymic generation of regulatory T cells in placental mammals mitigates
maternal–fetal conflict. Cell 2012; 150: 29–38. PMID:22770213,
DOI:10.1016/j.cell.2012.05.031
- 29. Josefowicz SZ, Niec RE, Kim HY, Treuting P,
Chinen T, Zheng Y, Umetsu DT, Rudensky AY: Extrathymically generated regulatory T cells
control mucosal TH2 inflammation. Nature 2012; 482: 395–399. PMID:22318520,
DOI:10.1038/nature10772
- 30. Iizuka-Koga M, Nakatsukasa H, Ito M, Akanuma T,
Lu Q, Yoshimura A: Induction and maintenance of regulatory T cells by transcription
factors and epigenetic modifications. J Autoimmun 2017; 83: 113–121. PMID:28709726,
DOI:10.1016/j.jaut.2017.07.002
- 31. Almeida AR, Legrand N, Papiernik M, Freitas AA:
Homeostasis of peripheral CD4+ T cells: IL-2R alpha and IL-2 shape a population of
regulatory cells that controls CD4+ T cell numbers. J Immunol 2002; 169: 4850–4860.
PMID:12391195, DOI:10.4049/jimmunol.169.9.4850
- 32. Bayer AL, Yu A, Adeegbe D, Malek TR: Essential
role for interleukin-2 for CD4+CD25+ T regulatory cell development during the neonatal
period. J Exp Med 2005; 201: 769–777. PMID:15753210,
DOI:10.1084/jem.20041179
- 33. Bayer AL, Yu A, Malek TR: Function of the IL-2R
for thymic and peripheral CD4+CD25+ Foxp3+ T regulatory cells. J Immunol 2007; 178:
4062–4071. PMID:17371960, DOI:10.4049/jimmunol.178.7.4062
- 34. Curotto de Lafaille MA, Lino AC, Kutchukhidze N,
Lafaille JJ: CD25– T cells generate CD25+Foxp3+ regulatory T cells by peripheral
expansion. J Immunol 2004; 173: 7259–7268. PMID:15585848,
DOI:10.4049/jimmunol.173.12.7259
- 35. Setoguchi R, Hori S, Takahashi T, Sakaguchi S:
Homeostatic maintenance of natural Foxp3+ CD25+ CD4+ regulatory T cells by interleukin
(IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med 2005; 201:
723–735. PMID:15753206, DOI:10.1084/jem.20041982
- 36. Burchill MA, Yang J, Vang KB, Farrar MA:
Interleukin-2 receptor signaling in regulatory T cell development and homeostasis. Immunol
Lett 2007; 114: 1–8. PMID:17936914, DOI:10.1016/j.imlet.2007.08.005
- 37. Li X, Liang Y, LeBlanc M, Benner C, Zheng Y:
Function of a Foxp3 cis-element in protecting regulatory T cell identity. Cell 2014; 158:
734–748. PMID:25126782, DOI:10.1016/j.cell.2014.07.030
- 38. Feng Y, Arvey A, Chinen T, van der Veeken J,
Gasteiger G, Rudensky AY: Control of the inheritance of regulatory T cell identity by a
cis element in the Foxp3 locus. Cell 2014; 158: 749–763. PMID:25126783,
DOI:10.1016/j.cell.2014.07.031
- 39. Polansky JK, Kretschmer K, Freyer J, Floess S,
Garbe A, Baron U, Olek S, Hamann A, von Boehmer H, Huehn J: DNA methylation controls Foxp3
gene expression. Eur J Immunol 2008; 38: 1654–1663. PMID:18493985,
DOI:10.1002/eji.200838105
- 40. Floess S, Freyer J, Siewert C, Baron U, Olek S,
Polansky J, Schlawe K, Chang HD, Bopp T, Schmitt E, Klein-Hessling S, Serfling E, Hamann
A, Huehn J: Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol 2007;
5: e38. PMID:17298177, DOI:10.1371/journal.pbio.0050038
- 41. Ohkura N, Hamaguchi M, Morikawa H, Sugimura K,
Tanaka A, Ito Y, Osaki M, Tanaka Y, Yamashita R, Nakano N, Huehn J, Fehling HJ, Sparwasser
T, Nakai K, Sakaguchi S: T cell receptor stimulation-induced epigenetic changes and Foxp3
expression are independent and complementary events required for Treg cell development.
Immunity 2012; 37: 785–799. PMID:23123060,
DOI:10.1016/j.immuni.2012.09.010
- 42. Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J,
Mathis D, Benoist C, Rudensky AY: Stability of the regulatory T cell lineage in vivo.
Science 2010; 329: 1667–1671. PMID:20929851, DOI:10.1126/science.1191996
- 43. Miyao T, Floess S, Setoguchi R, Luche H, Fehling
HJ, Waldmann H, Huehn J, Hori S: Plasticity of Foxp3+ T cells reflects promiscuous Foxp3
expression in conventional T cells but not reprogramming of regulatory T cells. Immunity
2012; 36: 262–275. PMID:22326580, DOI:10.1016/j.immuni.2011.12.012
- 44.Yadav M, Louvet C, Davini D, Gardner JM,
Martinez-Llordella M, Bailey-Bucktrout S, Anthony BA, Sverdrup FM, Head R, Kuster DJ,
Ruminski P, Weiss D, Von Schack D, Bluestone JA. Neuropilin-1 distinguishes natural and
inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med 2012; 209
: 1713–1722, S1711-1719, DOI:DOI:10.1084/jem.20120822
- 45.Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto
de Lafaille MA, Parkhurst CN, Xiong H, Dolpady J, Frey AB, Ruocco MG, Yang Y, Floess S,
Huehn J, Oh S, Li MO, Niec RE, Rudensky AY, Dustin ML, Littman DR, Lafaille JJ. Neuropilin
1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated
induced Foxp3+ T reg cells. J Exp Med 2012; 209 : 1723–1742, S1721,
DOI:DOI:10.1084/jem.20120914
- 46. Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H,
Litsa D, Bellucci R, Raderschall E, Canning C, Soiffer RJ, Frank DA, Ritz J: IL-2
regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent
mechanism and induces the expansion of these cells in vivo. Blood 2006; 108: 1571–1579.
PMID:16645171, DOI:10.1182/blood-2006-02-004747
- 47. Josefowicz SZ, Wilson CB, Rudensky AY: Cutting
edge: TCR stimulation is sufficient for induction of Foxp3 expression in the absence of
DNA methyltransferase 1. J Immunol 2009; 182: 6648–6652. PMID:19454658,
DOI:10.4049/jimmunol.0803320
- 48. Long M, Park SG, Strickland I, Hayden MS, Ghosh
S: Nuclear factor-kappaB modulates regulatory T cell development by directly regulating
expression of Foxp3 transcription factor. Immunity 2009; 31: 921–931. PMID:20064449,
DOI:10.1016/j.immuni.2009.09.022
- 49. Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC,
Greene MI, Tone M, Chen YH: Development of Foxp3+ regulatory T cells is driven by the
c-Rel enhanceosome. Immunity 2009; 31: 932–940. PMID:20064450,
DOI:10.1016/j.immuni.2009.10.006
- 50. Muto G, Kotani H, Kondo T, Morita R, Tsuruta S,
Kobayashi T, Luche H, Fehling HJ, Walsh M, Choi Y, Yoshimura A: TRAF6 is essential for
maintenance of regulatory T cells that suppress Th2 type autoimmunity. PLoS One 2013; 8:
e74639. PMID:24058613, DOI:10.1371/journal.pone.0074639
- 51. Sekiya T, Kashiwagi I, Yoshida R, Fukaya T,
Morita R, Kimura A, Ichinose H, Metzger D, Chambon P, Yoshimura A: Nr4a receptors are
essential for thymic regulatory T cell development and immune homeostasis. Nat Immunol
2013; 14: 230–237. PMID:23334790, DOI:10.1038/ni.2520
- 52. Sekiya T, Kashiwagi I, Inoue N, Morita R, Hori S,
Waldmann H, Rudensky AY, Ichinose H, Metzger D, Chambon P, Yoshimura A: The nuclear orphan
receptor Nr4a2 induces Foxp3 and regulates differentiation of CD4+ T cells. Nat Commun
2011; 2: 269. PMID:21468021, DOI:10.1038/ncomms1272
- 53.Sekiya T, Hibino S, Saeki K, Kanamori M,
Takaki S, Yoshimura A. Nr4a receptors regulate development and death of labile Treg
precursors to prevent generation of pathogenic self-reactive cells. Cell Rep 2018; 24 :
1627–1638.e1626, DOI:.DOI:10.1016/j.celrep.2018.07.008
- 54. Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez
J, Melamed R, Mathis D, Benoist C: Foxp3 transcription-factor-dependent and -independent
regulation of the regulatory T cell transcriptional signature. Immunity 2007; 27: 786–800.
PMID:18024188, DOI:10.1016/j.immuni.2007.09.010
- 55. Lin W, Haribhai D, Relland LM, Truong N, Carlson
MR, Williams CB, Chatila TA: Regulatory T cell development in the absence of functional
Foxp3. Nat Immunol 2007; 8: 359–368. PMID:17273171, DOI:10.1038/ni1445
- 56. Moran AE, Holzapfel KL, Xing Y, Cunningham NR,
Maltzman JS, Punt J, Hogquist KA: T cell receptor signal strength in Treg and iNKT cell
development demonstrated by a novel fluorescent reporter mouse. J Exp Med 2011; 208:
1279–1289. PMID:21606508, DOI:10.1084/jem.20110308
- 57. Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, Cui
K, Kanno Y, Roh TY, Watford WT, Schones DE, Peng W, Sun H, Paul WE, O’Shea JJ, Zhao K:
Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate
determination of differentiating CD4+ T cells. Immunity 2009; 30: 155–167. PMID:19144320,
DOI:10.1016/j.immuni.2008.12.009
- 58. Josefowicz SZ, Lu LF, Rudensky AY: Regulatory T
cells: mechanisms of differentiation and function. Annu Rev Immunol 2012; 30: 531–564.
PMID:22224781, DOI:10.1146/annurev.immunol.25.022106.141623
- 59. Cheng LE, Chan FK, Cado D, Winoto A: Functional
redundancy of the Nur77 and Nor-1 orphan steroid receptors in T-cell apoptosis. EMBO J
1997; 16: 1865–1875. PMID:9155013, DOI:10.1093/emboj/16.8.1865
- 60. Sekiya T, Kondo T, Shichita T, Morita R, Ichinose
H, Yoshimura A: Suppression of Th2 and Tfh immune reactions by Nr4a receptors in mature T
reg cells. J Exp Med 2015; 212: 1623–1640. PMID:26304965,
DOI:10.1084/jem.20142088
- 61. Chen J, López-Moyado IF, Seo H, Lio CW, Hempleman
LJ, Sekiya T, Yoshimura A, Scott-Browne JP, Rao A: NR4A transcription factors limit CAR T
cell function in solid tumours. Nature 2019; 567: 530–534. PMID:30814732,
DOI:10.1038/s41586-019-0985-x
- 62. Mognol GP, Spreafico R, Wong V, Scott-Browne JP,
Togher S, Hoffmann A, Hogan PG, Rao A, Trifari S: Exhaustion-associated regulatory regions
in CD8+ tumor-infiltrating T cells. Proc Natl Acad Sci USA 2017; 114: E2776–E2785.
PMID:28283662, DOI:10.1073/pnas.1620498114
- 63. Liu X, Wang Y, Lu H, Li J, Yan X, Xiao M, Hao J,
Alekseev A, Khong H, Chen T, Huang R, Wu J, Zhao Q, Wu Q, Xu S, Wang X, Jin W, Yu S, Wang
Y, Wei L, Wang A, Zhong B, Ni L, Liu X, Nurieva R, Ye L, Tian Q, Bian XW, Dong C:
Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature
2019; 567: 525–529. PMID:30814730, DOI:10.1038/s41586-019-0979-8
- 64. Hibino S, Chikuma S, Kondo T, Ito M, Nakatsukasa
H, Omata-Mise S, Yoshimura A: Inhibition of Nr4a receptors enhances antitumor immunity by
breaking Treg-mediated immune tolerance. Cancer Res 2018; 78: 3027–3040. PMID:29559474,
DOI:10.1158/0008-5472.CAN-17-3102
- 65.Lal G, Zhang N, van der Touw W, Ding Y, Ju W,
Bottinger EP, Reid SP, Levy DE, Bromberg JS. Epigenetic regulation of Foxp3 expression in
regulatory T cells by DNA methylation. J Immunol 2009; 182 , 259–273.
- 66. Nakatsukasa H, Oda M, Yin J, Chikuma S, Ito M,
Koga-Iizuka M, Someya K, Kitagawa Y, Ohkura N, Sakaguchi S, Koya I, Sanosaka T, Kohyama J,
Tsukada Y, Yamanaka S, Takamura-Enya T, Lu Q, Yoshimura A: Loss of TET proteins in
regulatory T cells promotes abnormal proliferation, Foxp3 destabilization and IL-17
expression. Int Immunol 2019; 31: 335–347. PMID:30726915,
DOI:10.1093/intimm/dxz008
- 67. Toker A, Engelbert D, Garg G, Polansky JK, Floess
S, Miyao T, Baron U, Düber S, Geffers R, Giehr P, Schallenberg S, Kretschmer K, Olek S,
Walter J, Weiss S, Hori S, Hamann A, Huehn J: Active demethylation of the Foxp3 locus
leads to the generation of stable regulatory T cells within the thymus. J Immunol 2013;
190: 3180–3188. PMID:23420886, DOI:10.4049/jimmunol.1203473
- 68. Nair VS, Oh KI: Down-regulation of Tet2 prevents
TSDR demethylation in IL2 deficient regulatory T cells. Biochem Biophys Res Commun 2014;
450: 918–924. PMID:24984151, DOI:10.1016/j.bbrc.2014.06.110
- 69. Wang L, Liu Y, Han R, Beier UH, Thomas RM, Wells
AD, Hancock WW: Mbd2 promotes foxp3 demethylation and T-regulatory-cell function. Mol Cell
Biol 2013; 33: 4106–4115. PMID:23979593, DOI:10.1128/MCB.00144-13
- 70. Yang R, Qu C, Zhou Y, Konkel JE, Shi S, Liu Y,
Chen C, Liu S, Liu D, Chen Y, Zandi E, Chen W, Zhou Y, Shi S: Hydrogen sulfide promotes
Tet1- and Tet2-mediated Foxp3 demethylation to drive regulatory T cell differentiation and
maintain immune homeostasis. Immunity 2015; 43: 251–263. PMID:26275994,
DOI:10.1016/j.immuni.2015.07.017
- 71. Yue X, Trifari S, Äijö T, Tsagaratou A, Pastor
WA, Zepeda-Martínez JA, Lio CW, Li X, Huang Y, Vijayanand P, Lähdesmäki H, Rao A: Control
of Foxp3 stability through modulation of TET activity. J Exp Med 2016; 213: 377–397.
PMID:26903244, DOI:10.1084/jem.20151438
- 72.Sasidharan Nair V., Song M. H. &Oh K. I.
Vitamin C facilitates demethylation of the Foxp3 enhancer in a Tet-dependent manner. J
Immunol 2016; DOI:10.4049/jimmunol.1502352
- 73. Yang BH, Hagemann S, Mamareli P, Lauer U,
Hoffmann U, Beckstette M, Föhse L, Prinz I, Pezoldt J, Suerbaum S, Sparwasser T, Hamann A,
Floess S, Huehn J, Lochner M: Foxp3+ T cells expressing RORγt represent a stable
regulatory T-cell effector lineage with enhanced suppressive capacity during intestinal
inflammation. Mucosal Immunol 2016; 9: 444–457. PMID:26307665,
DOI:10.1038/mi.2015.74
- 74. Someya K, Nakatsukasa H, Ito M, Kondo T, Tateda
K, Akanuma T, Koya I, Sanosaka T, Kohyama J, Tsukada Y, Takamura-Enya T, Yoshimura A:
Improvement of Foxp3 stability through CNS2 demethylation by TET enzyme induction and
activation. Int Immunol 2017; 29: 365–375. PMID:29048538,
DOI:10.1093/intimm/dxx049
- 75.Blaschke K, Ebata KT, Karimi MM,
Zepeda-Martínez JA, Goyal P, Mahapatra S, Tam A, Laird DJ, Hirst M, Rao A, Lorincz MC,
Ramalho-Santos M. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like
state in ES cells. Nature 2013; 500 : 222-226. DOI:10.1038/nature12362
- 76. Wu C, Chen Z, Dardalhon V, Xiao S, Thalhamer T,
Liao M, Madi A, Franca RF, Han T, Oukka M, Kuchroo V: The transcription factor musculin
promotes the unidirectional development of peripheral Treg cells by suppressing the TH2
transcriptional program. Nat Immunol 2017; 18: 344–353. PMID:28114290,
DOI:10.1038/ni.3667
- 77. Sasidharan Nair V, Song MH, Oh KI: Vitamin C
facilitates demethylation of the Foxp3 enhancer in a Tet-dependent manner. J Immunol 2016;
196: 2119–2131. PMID:26826239, DOI:10.4049/jimmunol.1502352
- 78. Takasato F, Morita R, Schichita T, Sekiya T,
Morikawa Y, Kuroda T, Niimi M, Yoshimura A: Prevention of allogeneic cardiac graft
rejection by transfer of ex vivo expanded antigen-specific regulatory T-cells. PLoS One
2014; 9: e87722. PMID:24498362, DOI:10.1371/journal.pone.0087722
- 79. Delacher M, Imbusch CD, Weichenhan D, Breiling A,
Hotz-Wagenblatt A, Träger U, Hofer AC, Kägebein D, Wang Q, Frauhammer F, Mallm JP, Bauer
K, Herrmann C, Lang PA, Brors B, Plass C, Feuerer M: Genome-wide DNA-methylation landscape
defines specialization of regulatory T cells in tissues. Nat Immunol 2017; 18: 1160–1172.
PMID:28783152, DOI:10.1038/ni.3799
- 80. Sharma A, Rudra D: Emerging functions of
regulatory T cells in tissue homeostasis. Front Immunol 2018; 9: 883. PMID:29887862,
DOI:10.3389/fimmu.2018.00883
- 81. DiSpirito JR, Zemmour D, Ramanan D, Cho J,
Zilionis R, Klein AM, Benoist C, Mathis D: Molecular diversification of regulatory T cells
in nonlymphoid tissues. Sci Immunol 2018; 3: eaat5861. PMID:30217811,
DOI:10.1126/sciimmunol.aat5861
- 82. Hayatsu N, Miyao T, Tachibana M, Murakami R,
Kimura A, Kato T, Kawakami E, Endo TA, Setoguchi R, Watarai H, Nishikawa T, Yasuda T,
Yoshida H, Hori S: Analyses of a mutant Foxp3 allele reveal BATF as a critical
transcription factor in the differentiation and accumulation of tissue regulatory T cells.
Immunity 2017; 47: 268–283.e9. PMID:28778586,
DOI:10.1016/j.immuni.2017.07.008
- 83. Danikowski KM, Jayaraman S, Prabhakar BS:
Regulatory T cells in multiple sclerosis and myasthenia gravis. J Neuroinflammation 2017;
14: 117. PMID:28599652, DOI:10.1186/s12974-017-0892-8
- 84. Lowther DE, Hafler DA: Regulatory T cells in the
central nervous system. Immunol Rev 2012; 248: 156–169. PMID:22725960,
DOI:10.1111/j.1600-065X.2012.01130.x
- 85.Jones A,A, Hawiger D. Peripherally induced
regulatory T cells: recruited protectors of the central nervous system against autoimmune
neuroinflammation. Front Immunol 2017; 8 : 532.
DOI:10.3389/fimmu.2017.00532
- 86. Bieber AJ, Kerr S, Rodriguez M: Efficient central
nervous system remyelination requires T cells. Ann Neurol 2003; 53: 680–684.
PMID:12731006, DOI:10.1002/ana.10578
- 87. Dombrowski Y, O’Hagan T, Dittmer M, Penalva R,
Mayoral SR, Bankhead P, Fleville S, Eleftheriadis G, Zhao C, Naughton M, Hassan R, Moffat
J, Falconer J, Boyd A, Hamilton P, Allen IV, Kissenpfennig A, Moynagh PN, Evergren E,
Perbal B, Williams AC, Ingram RJ, Chan JR, Franklin RJ, Fitzgerald DC: Regulatory T cells
promote myelin regeneration in the central nervous system. Nat Neurosci 2017; 20: 674–680.
PMID:28288125, DOI:10.1038/nn.4528
- 88. Liesz A, Suri-Payer E, Veltkamp C, Doerr H,
Sommer C, Rivest S, Giese T, Veltkamp R: Regulatory T cells are key cerebroprotective
immunomodulators in acute experimental stroke. Nat Med 2009; 15: 192–199. PMID:19169263,
DOI:10.1038/nm.1927
- 89. Liesz A, Kleinschnitz C: Regulatory T cells in
post-stroke immune homeostasis. Transl Stroke Res 2016; 7: 313–321. PMID:27030356,
DOI:10.1007/s12975-016-0465-7
- 90. Xu X, Li M, Jiang Y: The paradox role of
regulatory T cells in ischemic stroke. ScientificWorldJournal 2013; 2013: 1–8.
PMID:24288462, DOI:10.1155/2013/174373
- 91. Wimmer I, Zrzavy T, Lassmann H: Neuroinflammatory
responses in experimental and human stroke lesions. J Neuroimmunol 2018; 323: 10–18.
PMID:30196821, DOI:10.1016/j.jneuroim.2018.07.003
- 92. Shichita T, Ito M, Yoshimura A: Post-ischemic
inflammation regulates neural damage and protection. Front Cell Neurosci 2014; 8: 319.
PMID:25352781, DOI:10.3389/fncel.2014.00319
- 93. Jiang C, Kong W, Wang Y, Ziai W, Yang Q, Zuo F,
Li F, Wang Y, Xu H, Li Q, Yang J, Lu H, Zhang J, Wang J: Changes in the cellular immune
system and circulating inflammatory markers of stroke patients. Oncotarget 2017; 8:
3553–3567. PMID:27682880
- 94. Joy MT, Ben Assayag E, Shabashov-Stone D,
Liraz-Zaltsman S, Mazzitelli J, Arenas M, Abduljawad N, Kliper E, Korczyn AD, Thareja NS,
Kesner EL, Zhou M, Huang S, Silva TK, Katz N, Bornstein NM, Silva AJ, Shohami E,
Carmichael ST: CCR5 is a therapeutic target for recovery after stroke and traumatic brain
injury. Cell 2019; 176: 1143–1157.e13. PMID:30794775,
DOI:10.1016/j.cell.2019.01.044
- 95. Shichita T, Sugiyama Y, Ooboshi H, Sugimori H,
Nakagawa R, Takada I, Iwaki T, Okada Y, Iida M, Cua DJ, Iwakura Y, Yoshimura A: Pivotal
role of cerebral interleukin-17-producing γδT cells in the delayed phase of ischemic brain
injury. Nat Med 2009; 15: 946–950. PMID:19648929, DOI:10.1038/nm.1999
- 96. Shichita T, Hasegawa E, Kimura A, Morita R,
Sakaguchi R, Takada I, Sekiya T, Ooboshi H, Kitazono T, Yanagawa T, Ishii T, Takahashi H,
Mori S, Nishibori M, Kuroda K, Akira S, Miyake K, Yoshimura A: Peroxiredoxin family
proteins are key initiators of post-ischemic inflammation in the brain. Nat Med 2012; 18:
911–917. PMID:22610280, DOI:10.1038/nm.2749
- 97. Benakis C, Brea D, Caballero S, Faraco G, Moore
J, Murphy M, Sita G, Racchumi G, Ling L, Pamer EG, Iadecola C, Anrather J: Commensal
microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nat Med
2016; 22: 516–523. PMID:27019327, DOI:10.1038/nm.4068
- 98. Shichita T, Ito M, Morita R, Komai K, Noguchi Y,
Ooboshi H, Koshida R, Takahashi S, Kodama T, Yoshimura A: MAFB prevents excess
inflammation after ischemic stroke by accelerating clearance of damage signals through
MSR1. Nat Med 2017; 23: 723–732. PMID:28394332, DOI:10.1038/nm.4312
- 99. Ito M, Komai K, Mise-Omata S, Iizuka-Koga M,
Noguchi Y, Kondo T, Sakai R, Matsuo K, Nakayama T, Yoshie O, Nakatsukasa H, Chikuma S,
Shichita T, Yoshimura A: Brain regulatory T cells suppress astrogliosis and potentiate
neurological recovery. Nature 2019; 565: 246–250.
- 100. Stubbe T, Ebner F, Richter D, Engel OR, Klehmet
J, Royl G, Meisel A, Nitsch R, Meisel C, Brandt C: Regulatory T cells accumulate and
proliferate in the ischemic hemisphere for up to 30 days after MCAO. J Cereb Blood Flow
Metab 2013; 33: 37–47. PMID:22968321, DOI:10.1038/jcbfm.2012.128
- 101. Gadani SP, Walsh JT, Smirnov I, Zheng J, Kipnis
J: The glia-derived alarmin IL-33 orchestrates the immune response and promotes recovery
following CNS injury. Neuron 2015; 85: 703–709. PMID:25661185,
DOI:10.1016/j.neuron.2015.01.013
- 102. Lenglet S, Louiset E, Delarue C, Vaudry H,
Contesse V: Activation of 5-HT7 receptor in rat glomerulosa cells is associated with an
increase in adenylyl cyclase activity and calcium influx through T-type calcium channels.
Endocrinology 2002; 143: 1748–1760. PMID:11956157,
DOI:10.1210/endo.143.5.8817
- 103. Klein M, Bopp T: Cyclic AMP represents a crucial
component of Treg cell-mediated immune regulation. Front Immunol 2016; 7: 315.
PMID:27621729, DOI:10.3389/fimmu.2016.00315
- 104. Abeysinghe H, Phillips E, Chin-Cheng H, Beart P,
Roulston C: Modulating astrocyte transition after stroke to promote brain rescue and
functional recovery: emerging targets include Rho kinase. Int J Mol Sci 2016; 17: 288.
PMID:26927079, DOI:10.3390/ijms17030288
- 105. Liddelow SA, Guttenplan KA, Clarke LE, Bennett
FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin
A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM,
Stevens B, Barres BA: Neurotoxic reactive astrocytes are induced by activated microglia.
Nature 2017; 541: 481–487. PMID:28099414, DOI:10.1038/nature21029
- 106. Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki
T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H: Conditional ablation of
Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat
Med 2006; 12: 829–834. PMID:16783372, DOI:10.1038/nm1425
- 107. Xu Y, Meng C, Liu G, Yang D, Fu L, Zhang M, Zhang
Z, Xia H, Yao S, Zhang S: Classically activated macrophages protect against
lipopolysaccharide-induced acute lung injury by expressing amphiregulin in mice.
Anesthesiology 2016; 124: 1086–1099. PMID:26808632,
DOI:10.1097/ALN.0000000000001026
- 108. Kimura H, Schubert D: Schwannoma-derived growth
factor promotes the neuronal differentiation and survival of PC12 cells. J Cell Biol 1992;
116: 777–783. PMID:1530950, DOI:10.1083/jcb.116.3.777
- 109. Pang X, Qian W: Changes in regulatory T-cell
levels in acute cerebral ischemia. J Neurol Surg A Cent Eur Neurosurg 2017; 78: 374–379.
PMID:28320030, DOI:10.1055/s-0037-1599055
- 110.Dolati S, Ahmadi M, Khalili M, Taheraghdam
AA, Siahmansouri H, Babaloo Z, Aghebati-Maleki L, Jadidi-Niaragh F, Younesi V, Yousefi M.
Peripheral Th17/Treg imbalance in elderly patients with ischemic stroke. Neurol Sci 2018;
39 : 647–654, doi:DOI:10.1007/s10072-018-3250-4
- 111. Yan J, Read SJ, Henderson RD, Hull R, O’Sullivan
JD, McCombe PA, Greer JM: Frequency and function of regulatory T cells after ischaemic
stroke in humans. J Neuroimmunol 2012; 243: 89–94. PMID:22261543,
DOI:10.1016/j.jneuroim.2011.12.019
- 112. Swardfager W, Herrmann N, Andreazza AC, Swartz
RH, Khan MM, Black SE, Lanctôt KL: Poststroke neuropsychiatric symptoms: relationships
with IL-17 and oxidative stress. BioMed Res Int 2014; 2014: 1–6. PMID:25054133,
DOI:10.1155/2014/245210
- 113.Planas AM, Gómez-Choco M, Urra X, Gorina R,
Caballero M, Chamorro Á. Brain-derived antigens in lymphoid tissue of patients with acute
stroke. J Immunol 2012; 188 : 2156–2163, DOI:10.4049/jimmunol.1102289
- 114. Gu SC, Wang CD: Early selective serotonin
reuptake inhibitors for recovery after stroke: a meta-analysis and trial sequential
analysis. J Stroke Cerebrovasc Dis 2018; 27: 1178–1189. PMID:29276014,
DOI:10.1016/j.jstrokecerebrovasdis.2017.11.031
- 115. Chollet F, Tardy J, Albucher JF, Thalamas C,
Berard E, Lamy C, Bejot Y, Deltour S, Jaillard A, Niclot P, Guillon B, Moulin T, Marque P,
Pariente J, Arnaud C, Loubinoux I: Fluoxetine for motor recovery after acute ischaemic
stroke (FLAME): a randomised placebo-controlled trial. Lancet Neurol 2011; 10: 123–130.
PMID:21216670, DOI:10.1016/S1474-4422(10)70314-8
- 116. Fu Y, Zhang N, Ren L, Yan Y, Sun N, Li YJ, Han W,
Xue R, Liu Q, Hao J, Yu C, Shi FD: Impact of an immune modulator fingolimod on acute
ischemic stroke. Proc Natl Acad Sci USA 2014; 111: 18315–18320. PMID:25489101,
DOI:10.1073/pnas.1416166111
- 117. Subhramanyam CS, Wang C, Hu Q, Dheen ST:
Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin Cell Dev Biol
2019; S1084-9521(18)30089-2. PMID:31077796
- 118. Garretti F, Agalliu D, Lindestam Arlehamn CS,
Sette A, Sulzer D: Autoimmunity in Parkinson’s disease: the role of α-synuclein-specific T
cells. Front Immunol 2019; 10: 303. PMID:30858851,
DOI:10.3389/fimmu.2019.00303
- 119. Heppner FL, Ransohoff RM, Becher B: Immune
attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 2015; 16: 358–372.
PMID:25991443, DOI:10.1038/nrn3880
- 120. Duffy SS, Keating BA, Perera CJ, Moalem-Taylor G:
The role of regulatory T cells in nervous system pathologies. J Neurosci Res 2018; 96:
951–968. PMID:28488363, DOI:10.1002/jnr.24073
- 121. Sacramento PM, Monteiro C, Dias AS, Kasahara TM,
Ferreira TB, Hygino J, Wing AC, Andrade RM, Rueda F, Sales MC, Vasconcelos CC, Bento CA:
Serotonin decreases the production of Th1/Th17 cytokines and elevates the frequency of
regulatory CD4+ T-cell subsets in multiple sclerosis patients. Eur J Immunol 2018; 48:
1376–1388. PMID:29719048, DOI:10.1002/eji.201847525
- 122. Marei HE, Hasan A, Rizzi R, Althani A, Afifi N,
Cenciarelli C, Caceci T, Shuaib A: Potential of stem cell-based therapy for ischemic
stroke. Front Neurol 2018; 9: 34. PMID:29467713,
DOI:10.3389/fneur.2018.00034
- 123. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong
J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, Mathis D: Lean, but not obese, fat
is enriched for a unique population of regulatory T cells that affect metabolic
parameters. Nat Med 2009; 15: 930–939. PMID:19633656, DOI:10.1038/nm.2002
- 124. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J,
Shoelson SE, Benoist C, Mathis D: PPAR-γ is a major driver of the accumulation and
phenotype of adipose tissue Treg cells. Nature 2012; 486: 549–553. PMID:22722857,
DOI:10.1038/nature11132
- 125. Vasanthakumar A, Moro K, Xin A, Liao Y, Gloury R,
Kawamoto S, Fagarasan S, Mielke LA, Afshar-Sterle S, Masters SL, Nakae S, Saito H,
Wentworth JM, Li P, Liao W, Leonard WJ, Smyth GK, Shi W, Nutt SL, Koyasu S, Kallies A: The
transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of
adipose tissue-resident regulatory T cells. Nat Immunol 2015; 16: 276–285. PMID:25599561,
DOI:10.1038/ni.3085
- 126. Li C, DiSpirito JR, Zemmour D, Spallanzani RG,
Kuswanto W, Benoist C, Mathis D: TCR transgenic mice reveal stepwise, multi-site
acquisition of the distinctive fat-Treg phenotype. Cell 2018; 174: 285–299.e12.
PMID:29887374, DOI:10.1016/j.cell.2018.05.004
- 127. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL,
Cerletti M, Jang Y, Sefik E, Tan TG, Wagers AJ, Benoist C, Mathis D: A special population
of regulatory T cells potentiates muscle repair. Cell 2013; 155: 1282–1295. PMID:24315098,
DOI:10.1016/j.cell.2013.10.054
- 128. Villalta SA, Rosenthal W, Martinez L, Kaur A,
Sparwasser T, Tidball JG, Margeta M, Spencer MJ, Bluestone JA: Regulatory T cells suppress
muscle inflammation and injury in muscular dystrophy. Sci Transl Med 2014; 6: 258ra142.
PMID:25320234, DOI:10.1126/scitranslmed.3009925
- 129. Kuswanto W, Burzyn D, Panduro M, Wang KK, Jang
YC, Wagers AJ, Benoist C, Mathis D: Poor repair of skeletal muscle in aging mice reflects
a defect in local, interleukin-33-dependent accumulation of regulatory T cells. Immunity
2016; 44: 355–367. PMID:26872699, DOI:10.1016/j.immuni.2016.01.009
- 130. Panduro M, Benoist C, Mathis D: Treg cells limit
IFN-γ production to control macrophage accrual and phenotype during skeletal muscle
regeneration. Proc Natl Acad Sci USA 2018; 115: E2585–E2593. PMID:29476012,
DOI:10.1073/pnas.1800618115
- 131. Hofmann U, Beyersdorf N, Weirather J, Podolskaya
A, Bauersachs J, Ertl G, Kerkau T, Frantz S: Activation of CD4+ T lymphocytes improves
wound healing and survival after experimental myocardial infarction in mice. Circulation
2012; 125: 1652–1663. PMID:22388323,
DOI:10.1161/CIRCULATIONAHA.111.044164
- 132. Weirather J, Hofmann UD, Beyersdorf N, Ramos GC,
Vogel B, Frey A, Ertl G, Kerkau T, Frantz S: Foxp3+ CD4+ T cells improve healing after
myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res 2014;
115: 55–67. PMID:24786398, DOI:10.1161/CIRCRESAHA.115.303895
- 133. Tang TT, Yuan J, Zhu ZF, Zhang WC, Xiao H, Xia N,
Yan XX, Nie SF, Liu J, Zhou SF, Li JJ, Yao R, Liao MY, Tu X, Liao YH, Cheng X: Regulatory
T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res Cardiol 2012;
107: 232. PMID:22189560, DOI:10.1007/s00395-011-0232-6
- 134. Dobaczewski M, Xia Y, Bujak M, Gonzalez-Quesada
C, Frangogiannis NG: CCR5 signaling suppresses inflammation and reduces adverse remodeling
of the infarcted heart, mediating recruitment of regulatory T cells. Am J Pathol 2010;
176: 2177–2187. PMID:20382703, DOI:10.2353/ajpath.2010.090759
- 135. Garibaldi BT, D’Alessio FR, Mock JR, Files DC,
Chau E, Eto Y, Drummond MB, Aggarwal NR, Sidhaye V, King LS: Regulatory T cells reduce
acute lung injury fibroproliferation by decreasing fibrocyte recruitment. Am J Respir Cell
Mol Biol 2013; 48: 35–43. PMID:23002097, DOI:10.1165/rcmb.2012-0198OC
- 136. Mock JR, Garibaldi BT, Aggarwal NR, Jenkins J,
Limjunyawong N, Singer BD, Chau E, Rabold R, Files DC, Sidhaye V, Mitzner W, Wagner EM,
King LS, D’Alessio FR: Foxp3+ regulatory T cells promote lung epithelial proliferation.
Mucosal Immunol 2014; 7: 1440–1451. PMID:24850425, DOI:10.1038/mi.2014.33
- 137. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers
S, Yuan S, Treuting PM, Rudensky AY: A distinct function of regulatory T cells in tissue
protection. Cell 2015; 162: 1078–1089. PMID:26317471,
DOI:10.1016/j.cell.2015.08.021
- 138. Scharschmidt TC, Vasquez KS, Truong HA, Gearty
SV, Pauli ML, Nosbaum A, Gratz IK, Otto M, Moon JJ, Liese J, Abbas AK, Fischbach MA,
Rosenblum MD: A wave of regulatory T cells into neonatal skin mediates tolerance to
commensal microbes. Immunity 2015; 43: 1011–1021. PMID:26588783,
DOI:10.1016/j.immuni.2015.10.016
- 139. Sather BD, Treuting P, Perdue N, Miazgowicz M,
Fontenot JD, Rudensky AY, Campbell DJ: Altering the distribution of Foxp3 + regulatory T
cells results in tissue-specific inflammatory disease. J Exp Med 2007; 204: 1335–1347.
PMID:17548521, DOI:10.1084/jem.20070081
- 140. Scharschmidt TC, Vasquez KS, Pauli ML, Leitner
EG, Chu K, Truong HA, Lowe MM, Sanchez Rodriguez R, Ali N, Laszik ZG, Sonnenburg JL,
Millar SE, Rosenblum MD: Commensal microbes and hair follicle morphogenesis coordinately
drive Treg migration into neonatal skin. Cell Host Microbe 2017; 21: 467–477.e5.
PMID:28343820, DOI:10.1016/j.chom.2017.03.001
- 141. Ali N, Zirak B, Rodriguez RS, Pauli ML, Truong
HA, Lai K, Ahn R, Corbin K, Lowe MM, Scharschmidt TC, Taravati K, Tan MR, Ricardo-Gonzalez
RR, Nosbaum A, Bertolini M, Liao W, Nestle FO, Paus R, Cotsarelis G, Abbas AK, Rosenblum
MD: Regulatory T cells in skin facilitate epithelial stem cell differentiation. Cell 2017;
169: 1119–1129.e11. PMID:28552347, DOI:10.1016/j.cell.2017.05.002
- 142. Xu M, Pokrovskii M, Ding Y, Yi R, Au C, Harrison
OJ, Galan C, Belkaid Y, Bonneau R, Littman DR: c-MAF-dependent regulatory T cells mediate
immunological tolerance to a gut pathobiont. Nature 2018; 554: 373–377. PMID:29414937,
DOI:10.1038/nature25500
- 143. Schiering C, Krausgruber T, Chomka A, Fröhlich A,
Adelmann K, Wohlfert EA, Pott J, Griseri T, Bollrath J, Hegazy AN, Harrison OJ, Owens BM,
Löhning M, Belkaid Y, Fallon PG, Powrie F: The alarmin IL-33 promotes regulatory T-cell
function in the intestine. Nature 2014; 513: 564–568. PMID:25043027,
DOI:10.1038/nature13577