Metabolism in Human Pluripotent Stem Cells and Cardiomyocytes for Regenerative Therapy
Article ID: 2021-0015-IR

Details
Article ID: 2021-0015-IR
Pluripotent stem cells (PSCs), which include embryonic stem cells and induced pluripotent stem cells, have the potential for unlimited self-renewal and proliferation and the ability to differentiate into all three embryonic germ layers. Human PSCs (hPSCs) are used in drug discovery screening, disease models, and regenerative medicine. These cells maintain a transcriptional regulatory network based on a set of unique transcription factors to maintain their stem cell properties. Downstream of such transcriptional regulatory networks, various stem cell-specific metabolic programs are used to produce energy and metabolites as necessary. hPSCs and differentiated cells utilize different metabolic programs for self-renewal ability and maintenance of quiescence. Understanding the different metabolic features of hPSCs and differentiated cells can contribute to the development of technologies that are useful for regenerative medicine, such as the purification of differentiated cells. This review describes the unique metabolic programs active in hPSCs and their differences from somatic cells, with a focus on cardiomyocytes.
Pluripotent stem cells (PSCs), which include embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), are used in drug discovery screening, disease models, and regenerative medicine. Metabolism in PSCs greatly differs from that in differentiated somatic cells; therefore, understanding the metabolic features of PSCs can contribute to the development of regenerative medicine technologies. Here, we review the detailed metabolic features of PSCs and discuss their relevance to regenerative therapy with a focus on cardiomyocytes.
Rapidly proliferating cells show activated glycolysis even under conditions with sufficient oxygen (aerobic glycolysis).1 This phenomenon, which is observed in most cancer cells, was described by Otto Warburg and is known as the Warburg effect.2 Glycolysis is less energetically efficient than glucose oxidation via the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS). Glycolysis can occur in the absence of oxygen and results in ATP production at a much faster rate than that of oxygen-dependent respiration when glucose is abundant. This is a possible advantage of the Warburg effect, which produces sufficient biomass to enable rapid cell proliferation through the generation of nucleotides, nonessential amino acids, and lipids.
Aerobic glycolysis has been observed in human PSCs (hPSCs).3,4 In the inner cell mass of the blastocyst of the early embryo, glucose transporter 1 (GLUT1) and GLUT3, which transport glucose into the cell, are highly expressed and can take up glucose.5 The importance of these glycolytically dependent metabolic properties in blastocysts is supported by the fact that deficiencies in various glycolytic enzymes, such as hexokinase 2 and phosphofructokinase 1, result in preimplantation lethality.6 Cell division requires ATP as well as biosynthetic precursors derived from glycolysis, from the pentose phosphate pathway (PPP), and from the TCA cycle. Glucose 6-phosphate can be diverted from the glycolytic pathway to the PPP to produce ribose 5-phosphate for de novo nucleotide synthesis, which is also increased in hPSCs.4
Prioritization of glycolysis over mitochondrial oxidative metabolism may represent a mechanism for maintaining the integrity of hPSC genomes by reducing the levels of reactive oxygen species (ROS) to below those produced by OXPHOS, thereby limiting subsequent damage to nuclear and mitochondrial DNA, proteins, and lipids.7 Consequently, aerobic glycolysis is a common characteristic of hPSCs.
During reprogramming from somatic cells to human iPSCs (hiPSCs), metabolic profiles dramatically change from an oxidative state to a glycolytic state.8 Although the inhibition of glycolysis by 2-deoxy-D-glucose decreases the reprogramming efficiency of hiPSCs, stimulation of glycolysis by high glucose levels improves this efficiency.9 Studies comparing the gene expression profiles of hiPSCs and somatic cells revealed the upregulation of glycolytic genes with concomitant downregulation of pyruvate dehydrogenase in hiPSCs, thereby limiting the entry of glucose-derived pyruvate into the TCA cycle.4 Moreover, hexokinase 2 and pyruvate kinase M2 are directly regulated by the core pluripotency factor OCT4, and their overexpression limits human ESC differentiation.10,11 SOX2, OCT4, and NANOG, which are core pluripotent factors, promote glucose uptake and glycolysis in human ESCs by upregulating GLUT1 expression via direct binding to the enhancer of GLUT1.12 MYC also maintains a high glycolytic flux and pluripotency by sustaining the transcriptional activity of metabolic switch genes.3 During definitive endoderm and mesoderm differentiation, the overall MYC levels are reduced, metabolic switch genes are downregulated, and glycolytic flux is reduced. Similar to the processes occurring in cancer cells under hypoxic conditions, MYC and hypoxia inducible factor-1 may cooperate to induce metabolic shift during iPSC reprogramming.13
Pyruvate, a downstream metabolite of glycolysis, regulates hPSCs. Pyruvate can be metabolized to acetyl-coenzyme A (acetyl-CoA) by pyruvate dehydrogenase to enable entry and subsequent oxidation in the TCA cycle. Pyruvate can also be metabolized to lactate by lactate dehydrogenase, thereby enabling regeneration of nicotinamide adenine dinucleotide (NAD+) and supporting high glycolysis rates, and to oxaloacetate by pyruvate carboxylase to replenish the TCA cycle intermediates being utilized for anabolic pathways.14 In cancer cells, lactate is produced from pyruvate, whereas in hPSCs, acetyl-CoA is predominantly produced by aerobic glycolysis, which provides substrates for histone acetylation to maintain the pluripotency program.14 In summary, regulation of glycolytic flux plays an important role in the acquisition and maintenance of pluripotency (Fig. 1).
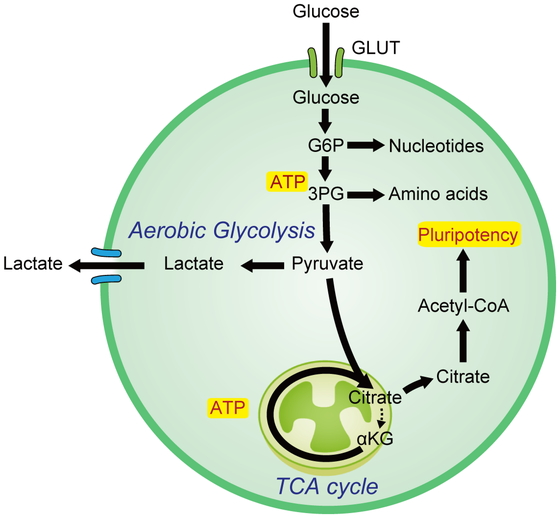
Aerobic glycolysis is a common feature of pluripotent stem cells (PSCs).
Acetyl-CoA, acetyl coenzyme A; αKG, alpha-ketoglutarate; ATP, adenosine triphosphate; GLUT, glucose transporter; G6P, glucose-6-phosphate; TCA, tricarboxylic acid; 3PG, 3-phosphoglyceric acid.
The mitochondria of somatic cells are elongated and have a network structure that extends throughout the cytoplasm; they also contain an intricate membrane structure known as cristae.15 Mitochondria are critical for maintaining cellular homeostasis, in part because of their canonical role as the major energy generator in the cell. Mitochondria play many roles in ROS production; calcium homeostasis; cellular signaling pathways; and the synthesis of metabolites such as fatty acids, amino acids, iron/sulfur clusters, pyrimidines, heme, and steroid hormones.16 It has become increasingly recognized that mitochondrial dynamics also significantly impact stem cell function and fate.17,18
In general, hPSCs have fewer mitochondria than do differentiated cells; hPSC mitochondria are generally small and globular, contain poorly developed and immature cristae, and are localized in the perinuclear region.9 hPSCs show high expression of the transporter uncoupling protein 2 (UCP2), which is localized in the inner mitochondrial membrane.19 UCP2 is highly expressed in specific cancer cells; in the presence of glucose, it suppresses the energy supply to the electron transport chain (ETC) by excreting oxaloacetate, maleic acid, and aspartic acid from mitochondria.20 In contrast, UCP2 activates the TCA cycle anaplerotically by promoting glutamine metabolism. As a result, UCP2 breaks the coupling between glycolysis and the OXPHOS pathway, thereby increasing the dependence of hPSCs on glycolysis metabolism.21 UCP2 expression decreases during differentiation, but overexpression of UCP2 causes a decrease in ROS and suppresses differentiation.19 Because the development of ROS promotes differentiation, a decrease in UCP2 may play an important role in differentiation via ROS.22 Collectively, these findings indicate that mitochondria exert functional and developmental roles in the metabolism of hPSCs.
TCA cycleThe mitochondrial TCA cycle is the central hub of energy metabolism and intersects many pathways involved in central carbon metabolism. The TCA cycle primarily oxidizes its major substrate, pyruvate, to CO2 to produce reducing factors and electron donors, including reduced nicotinamide adenine dinucleotide and flavin adenine dinucleotide, supplying the ETC and ATP synthesis.23 Many biosynthetic reactions use TCA intermediates; anaplerosis is the process of replenishing TCA cycle intermediates, primarily pyruvate and glutamine, which have been utilized for biosynthesis.24 By balancing energy production with cataplerosis to supply substrates for anabolic and post-translational protein modifications, the TCA cycle is important in regulating stem cell function and fate.25
Lipid metabolismLipids play important roles in maintaining cell homeostasis by acting as energy sources, signal transduction entities, and membrane components. Lipid metabolism represents a carefully adjusted balance between catabolism and anabolism and is highly dependent on the metabolic requirements of specific cellular states. Fatty acid oxidation involves the active transport of medium- and long-chain fatty acids to the mitochondria via a regulated carnitine palmitoyl transferase system. This is followed by β-oxidation to generate acetyl-CoA, which is fed to the TCA cycle, and NAD+, which donates its electrons to the ETC. In contrast, de novo fatty acid biosynthesis requires substrates from multiple metabolic pathways, such as acetyl-CoA, reducing factors, and ATP, to build essential fatty acids.
In mouse iPSCs, acetyl-CoA carboxylase and fatty acid synthase (FASN) are upregulated. These enzymes are involved in de novo fatty acid synthesis.26 Their pharmacological inhibition results in decreased reprogramming efficiency. Although de novo fatty acid synthesis is important for reprogramming into PSCs and for PSC survival, hPSCs do not utilize fatty acid oxidation.27 Knockdown or inhibition of FASN, which is the final enzyme involved in de novo fatty acid synthesis, induces apoptosis of undifferentiated hPSCs. Interestingly, hPSC-derived somatic cells, including cardiomyocytes (CMs), neurons, and hepatocytes, are unaffected by FASN inhibition. These data suggest that FASN inhibition is suitable for eliminating residual undifferentiated PSCs in clinical regenerative medicine approaches.
The dependence of hPSCs on glycolysis can be regulated under various culture conditions. hPSCs utilize glycolysis to a greater or lesser extent depending on whether they are cultured in the absence or presence, respectively, of feeder cells.28 PPP, which diverts from the glycolytic pathway, can support the regeneration of reduced nicotinamide adenine dinucleotide phosphate (NADPH). NADPH is essential for the regeneration of reduced glutathione from glutathione disulfide; NADPH also functions as a cofactor in many biosynthetic reactions. The metabolic reprogramming induced by lipid deficiency increases oxidative PPP to support NADPH regeneration, increased glutamine consumption, and fatty acid biosynthesis. This is consistent with the importance of glutaminolysis and PPP in lipid biosynthesis in hPSCs.4
Amino acid metabolismNutritional factors in the culture medium, particularly the amino acid composition, are important for maintaining PSCs. Mouse ESCs have a much higher requirement for threonine than for other amino acids that supply carbon to the one-carbon metabolism pathway.29 Threonine dehydrogenase and the downstream enzymes involved in threonine metabolism are highly expressed in mouse ESCs and are rapidly downregulated during differentiation. Indeed, inhibition of threonine dehydrogenase or complete removal of threonine from the culture medium results in a loss of stemness, reduced proliferation, and apoptosis,29 whereas L-threonine supplementation supports the induction of pluripotency through nuclear reprogramming.30 The metabolism of threonine and the accompanying cleavage of glycine supply a folic acid-dependent 1-carbon pool, producing S-adenosylmethionine (SAM) in addition to nucleic acids.31 SAM is used for histone methylation, which is necessary for maintaining pluripotency. In contrast, hPSCs have high methionine demands to supply SAM.32
Like other highly proliferating cells, hPSCs are dependent on glutamine.33,34,35 Glutamine is a non-essential amino acid that is converted to glutamate by glutaminase in the nitrogen-donating reaction of nucleotide synthesis.33 Glutamine also enters the TCA cycle through glutamate, followed by conversion to α-ketoglutarate (αKG) by glutamate dehydrogenase (glutaminolysis). Glutaminolysis is necessary not only for driving the TCA cycle for ATP production but also for synthesizing reduced glutathione (GSH) in hPSCs.33 Glutamine supports the stability of OCT4 in hPSCs by maintaining GSH levels.35 αKG is an important cofactor for αKG-dependent dioxygenase enzymes, including Jumonji domain-containing histone demethylase and ten-eleven translocation enzymes.36 The importance of αKG in regulating pluripotency by epigenetic modification has been previously demonstrated in several studies.36,37
Tryptophan (TRP) metabolism plays an important role in proliferation while maintaining the pluripotency of hPSCs.38 Kynurenine (KYN), a TRP metabolite generated by the enzyme indoleamine 2,3-dioxygenase 1 (IDO1), is a ligand for the aryl hydrocarbon receptor. IDO1, KYN, and aryl hydrocarbon receptor are required for the self-renewal of hPSCs. KYN synthesis is required to maintain the undifferentiated state of hPSCs, and KYN degradation is required for the differentiation of hPSCs into the ectoderm. In addition, levels of intracellular and extracellular KYN are reduced under TRP supplementation conditions, whereas levels of N-formylkynurenine, an upstream metabolite of KYN, are increased, thereby contributing to growth promotion.39 Taken together, TRP is indispensable for the survival and proliferation of hPSCs.
Multiple clinical trials to evaluate the use of hPSCs in regenerative medicine are currently underway. However, the risk of tumorigenicity is a major limitation of their use in clinical situations. An innovative method was developed to eliminate undifferentiated PSCs and thereby purify differentiated cells by altering the culture conditions based on the understanding of the metabolic signature in hPSCs.27,29,32,33,39,40 Understanding the unique metabolic signatures of hPSCs will enable advances in regenerative medicine and drug discovery (Fig. 2).
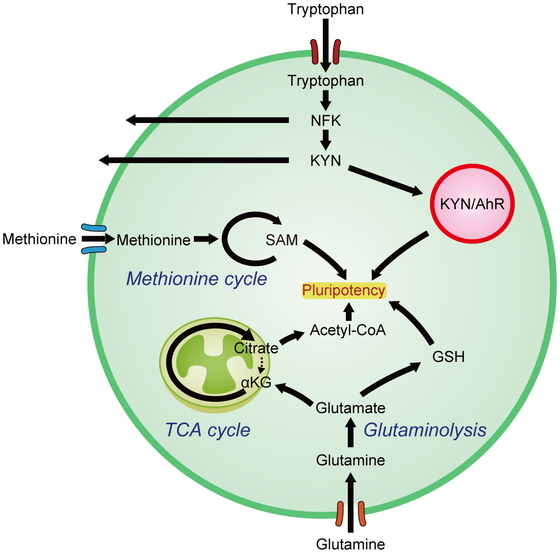
Amino acid metabolism plays an important role in maintaining pluripotency and proliferation of PSCs.
AhR, aryl hydrocarbon receptor; NFK, N-formylkynurenine; GSH, reduced glutathione; KYN, kynurenine; SAM, S-adenosylmethionine.
hiPSC-derived cardiomyocytes (hiPSC-CMs) are immature and resemble fetal cells in terms of their size, sarcomere structure, contractile activity, electrophysiological activity, mitochondrial structure, and metabolism.41 Adult mature CMs primarily rely on fatty acid oxidation for energy production, whereas fetal CMs primarily rely on glycolysis and lactic acid oxidation.
Differentiation from hiPSCs to hiPSC-CMs involves a transition from aerobic glycolysis to mitochondrial oxidation.42 This difference in metabolism can be used to purify hiPSC-CMs after differentiation from hiPSCs. Because undifferentiated hiPSCs and non-CMs derived from hiPSCs predominantly depend on glucose and glutamine metabolism, cell death results if they are deprived of glucose and glutamine. In contrast, hiPSC-CMs consume lactate to fuel the TCA cycle and generate energy. hiPSC-CMs can survive under glucose- and glutamine-deficient conditions in the presence of lactate.33,40 In lactate-supplemented medium from which glucose and glutamine are removed, undifferentiated hiPSCs and non-CMs derived from hiPSCs cannot survive, whereas hiPSC-CMs can survive, thereby enabling purification of hiPSC-CMs. This method is inexpensive, simple, and expandable, and is particularly promising for cardiac regeneration treatment using hiPSC-CMs.43
However, the immaturity of hiPSC-CMs may limit their application in regenerative medicine, drug discovery, and disease modeling. Long-term culture of hiPSC-CMs improves cell maturation, but hiPSC-CMs subjected to long-term culture reportedly cannot attain the maturation level of adult CMs.44 Several methods have been reported to promote maturation, including culture media, mechanical stress, electrical stimulation, and thyroid hormone.45,46,47 Metabolism is an important factor affecting cell properties and can be further utilized to promote the maturation of hiPSC-CMs.
Switching energy sources not only increases mitochondrial number and metabolism, but also promotes morphological, structural, and physiological maturation. In contrast, culturing hiPSC-CMs in high-glucose medium promotes nucleotide biosynthesis and inhibits structural and functional maturation.48 This is because of decreased glucose uptake and increased nucleotide deficiency in the heart during late pregnancy and early postnatal life. Recently, an estrogen-related receptor gamma agonist was shown to induce hiPSC-CMs to promote maturation in terms of cardiac gene expression, morphology, and function.49,50 Overall, these findings underscore the importance of metabolism and the need to optimize culture conditions to promote the maturation of hiPSC-CMs (Fig. 3).
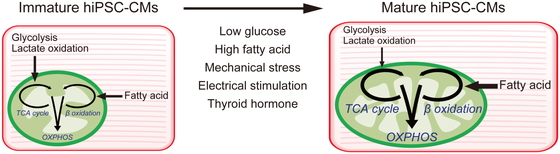
Metabolic switch enhances human induced pluripotent stem cell-derived cardiomyocyte (hiPSC-CM) maturation.
OXPHOS, oxidative phosphorylation.
In this review, we detail the metabolic features of PSCs and their effects on the properties of PSCs. Although hiPSC-derived somatic cells are currently being clinically applied in regenerative medicine, various limitations, particularly safety concerns, require further analysis. Exploiting the metabolic characteristics governing cells at different stages may serve as an effective strategy for mitigating these issues.
This work was supported by Projects for Technological Development, Research Center Network for Realization of Regenerative Medicine by Japan, Japan Agency for Medical Research and Development (AMED) grant 20bm0404023h0003 to S.T.; partly supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI 20H03768 to S.T.; and partly supported by Grant-in-Aid for JSPS Fellows 21J21186 to T.C.U.
S.T. is an advisor of Heartseed, Inc., and owns equity in Heartseed, Inc. T.C.U. has no conflicts of interest to disclose.