Molecular Basis of Hereditary Hair Diseases
Article ID: 2023-0007-IR

Details
Article ID: 2023-0007-IR
The hair follicle is an appendage of the skin that undergoes hair cycles throughout life. Recently, numerous genes expressed in the hair follicles have been identified, and variants in some of these genes are now known to underlie hereditary hair diseases in humans. Hereditary hair diseases are classified into non-syndromic and syndromic forms. In the Japanese population, the non-syndromic form of autosomal recessive woolly hair, which is caused by founder pathogenic variants in the lipase H (LIPH) gene, is the most prevalent hereditary hair disease. In addition, other types of hereditary hair diseases are known in Japan, such as Marie-Unna hereditary hypotrichosis, hypohidrotic ectodermal dysplasia, and tricho–rhino–phalangeal syndrome. To ensure correct diagnoses and appropriate patient care, dermatologists must understand the characteristics of each hair disorder. Elucidation of the molecular basis of hereditary hair diseases can directly tell us which genes are crucial for morphogenesis and development of hair follicles in humans. Therefore, continuation of “wet laboratory” research for these diseases remains important. To date, several syndromic forms of hereditary hair diseases have been approved as designated intractable diseases in Japan. As part of our efforts in the Project for Research on Intractable Diseases through the Ministry of Health, Labour, and Welfare of Japan, we anticipate that more hereditary hair diseases be recognized as designated intractable diseases in the future, which will be to the benefit of the affected individuals.
Mammalian hair follicle (HF), an appendage of the skin, has self-renewal ability and undergoes hair cycles throughout the postnatal life that consist of anagen (growing), catagen (regressing), and telogen (resting) phases.1 This wonderful capacity is maintained by HF epithelial stem cells, located in the bulge portion of the outer root sheath, and by the dermal papilla cells (Fig. 1).2 During the anagen phase, daughter cells derived from the bulge portion move into the matrix of the HF (Fig. 1). Then the matrix cells, which are stimulated by the dermal papilla cells, subsequently amplify and differentiate into distinct layers such as the hair shaft and the inner root sheath (IRS) (Fig. 1).3 These cell layers are differentially keratinized at the keratinizing zone, leading to the formation of a rigid structure (Fig. 1). The IRS molds to the hair shaft and supports its growth. After the hair shaft is completely keratinized, the IRS spontaneously collapses. Because the telogen HF does not have the IRS, the hair shaft can be easily plucked. In healthy scalp skin, the respective durations of the anagen, catagen, and telogen phases are 2–6 years, 1–2 weeks, and 2–3 months. Typically, about 85%–90% of scalp HFs are in the anagen phase, while 10%–15% of the HFs are in either the catagen or telogen phases. Notably, this ratio can change dramatically under some conditions. For example, in people suffering from COVID-19 infection, the number of telogen HFs eventually increases and may lead to temporary hair loss.4 A similar phenomenon can frequently be observed in women soon after childbirth as the result of a condition known as telogen effluvium.

Schematic representation of anagen hair follicle.
Cells derived from the bulge portion move into the matrix region (arrows) and subsequently differentiate into each component of the hair follicle.
Until recently, little was known about the types of molecules that are expressed in HFs, and the molecular mechanisms responsible for the control of hair cycles were not well defined. Recent advances in molecular genetics have enabled us to identify numerous genes that are expressed in HFs. Furthermore, in the past two decades, gradual advances have revealed that variants in some of these genes underlie hereditary hair diseases in humans.5,6,7 These findings have helped to identify the genes that are crucial for normal morphogenesis and/or development of human HFs. When compared with other mammals, hair coverage may not be considered essential for humans. However, hair loss disorders can severely affect the quality of life in affected individuals, and most of the diseases are intractable.
Hereditary hair diseases are largely classified into non-syndromic and syndromic forms. In the non-syndromic forms, affected individuals only show hair symptoms. In contrast, those with syndromic forms can show various abnormalities in addition to hair symptoms, such as hypohidrosis, nail dystrophy, ichthyosis, hypodontia, and cleft lip/palate. This review article introduces hereditary hair diseases from non-syndromic and syndromic groups, mainly in the Japanese population. In addition, the review discusses our recent achievements in the Project for Research on Intractable Diseases by the Ministry of Health, Labour, and Welfare of Japan regarding hereditary hair diseases.
Several types of non-syndromic hereditary hair diseases are known. The following discussion describes the clinical features and molecular basis of each disorder (Table 1).
| Disease | Inheritance pattern |
Gene |
|---|---|---|
| Woolly hair | AR | LIPH, LPAR6, C3orf52, KRT25 |
| AD | KRT74, KRT71, KRT25 | |
| Monilethrix | AD | KRT81, KRT83, KRT86 |
| AR | DSG4 | |
| Marie-Unna hereditary hypotrichosis | AD | U2HR |
| Hypotrichosis simplex | AD | CDSN, APCDD1, RPL21, SNRPE |
| AR | LSS |
AR, autosomal recessive; AD, autosomal dominant.
Woolly hair (WH) is characterized by tightly curled hair that stops growing at a few inches. Most affected individuals with WH also show sparse scalp hair (hypotrichosis) (Fig. 2). Observation of WH by electron microscopy reveals irregularly curved hairs, as well as a rough formation of the cuticle structure.5 Hereditary WH can be divided into non-syndromic and syndromic forms. Of these, affected individuals with syndromic forms can exhibit various abnormalities, such as palmoplantar keratoderma and cardiomyophathy.8 The syndromic forms of WH are extremely rare; most patients with WH exhibit the non-syndromic forms, which can show either autosomal dominant or recessive inheritance patterns.
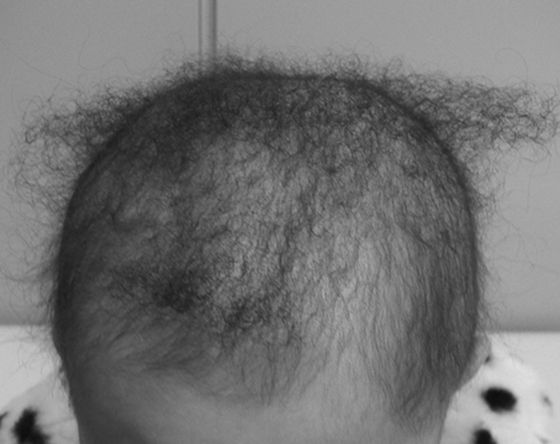
Clinical features of woolly hair.
The patient was a 3-year-old Japanese girl with autosomal recessive woolly hair caused by bi-allelic founder variants in the LIPH gene.
The major causative genes for non-syndromic forms of autosomal recessive WH (ARWH) are lipase H (LIPH) and lysophosphatidic acid receptor 6 (LPAR6).9,10 The LIPH gene encodes a cell membrane-associated phospholipase A1, known as PA-PLA1α, that produces 2-acyl-lysophosphatidic acid (2-acyl-LPA) from phosphatidic acid.11 The LPAR6 gene encodes a G-protein-coupled receptor named LPA6, which has been demonstrated to be a specific receptor for 2-acyl-LPA (Fig. 3).12 Most recently, bi-allelic loss-of-function variants in C3orf52 (TTMP) gene have been reported to underlie ARWH.13 In vitro studies in cultured cells have shown that C3orf52 protein is expressed at the cell membrane and supports the function of PA-PLA1α (Fig. 3).13 Importantly, PA-PLA1α, LPA6, and C3orf52 proteins are co-expressed at the IRS of human HFs.9,10,13 Therefore, it is now recognized that the LIPH, LPAR6, and C3orf52 genes are functionally related in a signaling pathway of lipid mediators and play crucial roles in development of the IRS and hair growth in humans.

Schematic representation of functional relationships between causative genes/molecules for hereditary hair diseases.
Gray arrows indicate an enzymatic digestion. PA, phosphatidic acid; LPA, lysophosphatidic acid.
Non-syndromic forms of autosomal dominant WH are known to be caused by pathogenic variants in keratin 74 (KRT74), keratin 71 (KRT71), or keratin 25 (KRT25) genes,14,15,16 all of which encode an IRS-specific epithelial keratin. Of these, KRT25 has also been reported to be a causative gene for ARWH.17 In mouse HFs, it has been demonstrated that PA-PLA1α/LPA/LPA6 signaling leads to activation of epithelial growth factor receptor (EGFR) and regulates expression of the IRS-specific keratins.18 The same molecular mechanism is predicted to exist in human HFs based on the phenomenon that EGFR kinase inhibitors can cause WH and hair loss as adverse events in cancer patients.
It is noteworthy that ARWH resulting from pathogenic variants in the LIPH gene is apparently the most prevalent hereditary hair disease in the Japanese population.19,20 To date, two founder variants have been known: c.736T>A (p.C246S) and c.742C>A (p.H248N), both of which are located in exon 6 of the LIPH gene. Because of the high frequency of these variants, there are an estimated 10,000 Japanese patients with ARWH caused by LIPH gene variants.
MonilethrixMonilethrix is a hair shaft anomaly that is characterized by periodic changes in hair diameter.21 In addition to scalp hairs, facial and body hairs can also be affected. The hairs of monilethrix are fragile and can frequently cause follicular plugs of the hair shaft, leading to follicular papules and perifollicular erythema. The disease can show either autosomal dominant or autosomal recessive inheritance patterns. Of these, the autosomal dominant forms of monilethrix are caused by pathogenic variants in keratin 81 (KRT81), keratin 83 (KRT83), or keratin 86 (KRT86) genes.22,23 These keratins are type II hair keratins that are predominantly expressed in the keratinizing zone of the hair shaft cortex.24 Because these hair keratins are also expressed in the nail units, affected individuals can show nail dystrophy, which is observed as spoon nails in most cases. The autosomal recessive forms of the disease, which are less frequent than the autosomal dominant forms, are known to be caused by pathogenic variants in desmoglein 4 (DSG4) gene.25 Importantly, DSG4 is the only desmoglein expressed in the hair shaft.26 The mechanisms of how pathogenic variants in the hair keratin and DSG4 genes result in a similar phenotype may be explained by the overlapped expression patterns in the hair shaft cortex and the close interaction between keratins and desmosomes within the cytoplasm (Fig. 3).
Marie-Unna hereditary hypotrichosisMarie-Unna hereditary hypotrichosis (MUHH) is an autosomal dominant inheritance disorder that is characterized by hypotrichosis on the scalp resembling androgenetic alopecia and irregularly twisted hairs called wiry hair. Eyebrows and eyelashes are also sparse in most cases. The disease had previously been mapped to the hairless (HR) gene locus on chromosome 8p21.3.27 However, no pathogenic variants were identified in exons or exon–intron boundaries of the HR gene. Strikingly, a Chinese study has found that there are several small open reading frames (ORFs) within the promoter region of the HR gene, and pathogenic variants in one of these ORFs were subsequently identified as U2HR in Chinese families with MUHH, which showed completely segregated phenotypes.28 The U2HR gene encodes a small peptide that is composed of only 34 amino acid residues. Importantly, U2HR protein has been shown to regulate expression of the HR protein (Fig. 3), and loss of function of U2HR is believed to lead to abnormalities in hair cycles and hair shaft differentiation.28
Hypotrichosis simplexHypotrichosis simplex (HS) is a non-syndromic hereditary hair disease that is diagnosed when the affected individuals do not show any characteristic hair shaft anomalies. HS can show either autosomal dominant or recessive inheritance patterns. To date, a total of four genes (CDSN, APCDD1, RPL21, and SNRPE) have been reported to be causative for the autosomal dominant forms,29,30,31,32 whereas pathogenic variants in any of these genes have not been identified in Japanese patients with HS to date. Recently, bi-allelic loss-of-function variants in the LSS gene have been identified in families with autosomal recessive HS in several populations, including the Japanese.33,34,35 The hypotrichosis phenotypes caused by the LSS gene variants are more severe than those of autosomal dominant forms; namely, most patients with LSS gene variants only have short vellus hairs on the scalp that are easily plucked (Fig. 4), suggesting that the hair cycles are extraordinary shortened.34 The LSS gene encodes lanosterol synthase, which is an enzyme in a cholesterol synthesis pathway.33 Interestingly, in addition to HS, variants in the LSS gene have been reported to underlie other conditions including non-syndromic congenital cataract, alopecia with intellectual disability, hypotrichosis with growth abnormality, and palmoplantar keratoderma with congenital alopecia syndrome.36,37,38,39 At present, however, there are no clear genotype–phenotype correlations regarding LSS gene variants. In the future, further studies will be required to characterize each LSS gene variant toward unraveling the mechanisms that lead to such a wide range of phenotypic variations.

Clinical features of autosomal recessive hypotrichosis simplex.
The patient was a 5-year-old Japanese boy who carried bi-allelic variants in the LSS gene.
It is known that more than 200 syndromes can show particular hair symptoms. Identification of causative genes for these diseases can contribute to elucidating the molecular basis of morphogenesis and/or development of not only HFs, but also other organs. The following discussion describes representative syndromic forms of hereditary hair diseases (Table 2).
| Disease | Inheritance pattern |
Gene |
|---|---|---|
| Hypohidrotic ectodermal dysplasia (HED) | XR | EDA |
| AD/AR | EDAR, EDARADD | |
| ED and immunodeficiency | XR | IKBKG |
| Odonto–onycho–dermal dysplasia | AR | WNT10A |
| Tricho–rhino–phalangeal syndrome | AD | TRPS1 |
| TP63-associated ED syndromes | AD | TP63 |
| Hypotrichosis with juvenile macular dystrophy | AR | CDH3 |
| ED, ectrodactyly, and macular dystrophy syndrome | ||
| Clouston syndrome | AD | GJB6 |
| Cleft lip/palate–ED syndrome | AR | NECTIN1 |
| Netherton syndrome | AR | SPINK5 |
| Keratitis–ichthyosis–deafness syndrome | AD | GJB2 |
| AR | AP1B1 | |
| Ichthyosis follicularis–atrichia–photophobia syndrome | XR | MBTPS2 |
| AD | SREBF1 |
XR, X-linked recessive; AD, autosomal dominant; AR, autosomal recessive.
Hypohidrotic ectodermal dysplasia (HED) is a major form of ED and is characterized by decreased sweating (hypohidrosis), sparse teeth (hypodontia), and hypotrichosis. Affected individuals of HED typically show abnormal facial features such as saddle nose, periorbital pigmentation, and low-set ears. In addition, they frequently have dry skin and atopic dermatitis most likely caused by hypohidrosis-induced abnormal barrier function of the epidermis. Most cases of HED show X-linked recessive inheritance pattern, while families with HED showing either autosomal dominant or recessive inheritance traits also exist. The X-linked recessive form of HED is caused by pathogenic variants in the ectodysplasin (EDA) gene,40 and the autosomal dominant/recessive forms of the disease result from pathogenic variants in either ectodysplasin A receptor (EDAR) or EDAR-associated death domain (EDARADD) genes,41,42 respectively. The EDA gene encodes several isoforms through alternative splicing events, of which EDA-A1 is the main isoform belonging to the tumor necrosis factor (TNF) ligand superfamily. EDA-A1 is secreted and functions as a ligand of EDAR, which is a member of the TNF receptor superfamily. EDARADD interacts with the intracellular domain of EDAR and supports its function as an adaptor protein. As such, EDA-A1, EDAR, and EDARADD are directly associated with each other in a signaling pathway known as EDAR signaling. This signaling pathway activates the transcription factor NF-κB, which subsequently move into the nucleus and regulate many genes responsible for ectodermal development (Fig. 3).43 It is also known that pathogenic variants in genes encoding the downstream molecules of the EDAR signaling, such as the IKBKG (NEMO) gene, underlie ED and immunodeficiency.44
In some publications, it is stated that WNT10A is an additional causative gene for HED.45 Indeed, recessive inherited variants in the WNT10A gene can show hypohidrosis, hypodontia, and hypotrichosis, similar to HED. The phenotypes resulting from WNT10A gene variants, however, are apparently distinct from HED; namely, affected individuals with WNT10A gene variants also show dystrophic nails and palmoplantar keratoderma, whereas they do not have abnormal facial features. The disease caused by WNT10A gene variants is officially named odonto-onycho-dermal dysplasia (OODD).46 The WNT10A gene encodes a ligand of WNT signaling that is known to be functionally related with EDAR signaling.47 It can explain the phenotypic overlaps between HED and OODD.
Tricho–rhino–phalangeal syndromeTricho–rhino–phalangeal syndrome (TRPS) is an autosomal dominant inheritance disorder that is characterized by hypotrichosis and craniofacial and skeletal abnormalities. Affected individuals show characteristic facial features including a pear-shaped nose and thin upper lip. The skeletal abnormalities mainly involve fingers and toes, which typically show brachydactyly and clinodactyly. Examination under X-ray reveals cone-shaped epiphyses.48 In cases with mild skeletal abnormalities in which the attending physician fails to carefully examine the patient’s face, misdiagnosis of a non-syndromic form of hypotrichosis may occur. The traits of this disease indicate that in addition to observing scalp hairs, the entire body should also be examined for any other abnormalities when the patient is suspected of having a hereditary hair disease.
TRPS is caused by mono-allelic loss-of-function variants in the TRPS1 gene.49 The TRPS1 gene encodes a transcription factor that is important for development of HFs and bones. To date, many Japanese cases of TRPS have been reported in the literature; therefore, TRPS may be the most prevalent syndromic form of hereditary hair disease in the Japanese population.
TP63 gene-associated ED syndromesTP63 gene encodes p63, which is a major transcription factor in epithelia including the epidermis and skin appendages. Mono-allelic pathogenic variants in the TP63 gene are known to underlie various autosomal dominant inheritance ED syndromes, such as ectrodactyly–ED–cleft lip/palate (EEC) syndrome, acro-dermato-ungual-lacrimal-tooth (ADULT) syndrome, ankyloblepharon–ectodermal defects–cleft lip/palate (AEC) syndrome, and Rapp-Hodgkin syndrome.50,51,52,53 Common clinical features among these diseases are hypotrichosis with pili-torti, nail dystrophy, hypohidrosis and hypodontia, lacrimal duct obstruction, and cleft lip/palate. In EEC and ADULT syndromes, affected individuals also show split hand/foot malformation (ectrodactyly) and/or syndactyly. There are clear genotype–phenotype correlations, namely, variants in the DNA-binding domain lead to either EEC or ADULT syndrome, whereas those in the SAM domain underlie either AEC or Rapp-Hodgkin syndrome.
It has gradually been disclosed that several causative genes (including CDH3, GJB6, and NECTIN1) for syndromic forms of hereditary hair disorders are direct transcriptional targets of p63 (Fig. 3).54,55,56 Recessive inheritance variants in the CDH3 gene encoding P-cadherin cause either hypotrichosis with macular dystrophy or ectodermal dysplasia, ectrodactyly, and macular dystrophy syndrome.57,58 Dominant inheritance variants in the GJB6 gene encoding connexin 30 underlie Clouston syndrome, which is characterized by hypotrichosis, nail dystrophy, and palmoplantar keratoderma.59 Finally, recessive inheritance variants in the NECTIN1 gene, which encodes a component of adherens junctions, are known to be causative for cleft lip/palate-ectodermal dysplasia syndrome, which is characterized by cleft lip/palate, hypotrichosis, and syndactyly.60 All of these diseases can show partially overlapped phenotypes with those of TP63 gene-associated ED.
Syndromic hair disorders with congenital ichthyosisCongenital ichthyosis can be largely classified into non-syndromic and syndromic forms.61 Among the syndromic forms of ichthyosis, there are several diseases showing hair symptoms, including Netherton syndrome, keratitis–ichthyosis–deafness (KID) syndrome, and ichthyosis follicularis–atrichia–photophobia (IFAP) syndrome.
Netherton syndrome is an autosomal recessive disorder characterized by congenital ichthyosis, atopic disposition, and hypotrichosis with a specific hair shaft anomaly known as trichorrhexis invaginata (bamboo hair), which is caused by bi-allelic pathogenic variants in the SPINK5 gene encoding lympho-epithelial Kazal-type-related inhibitor (LEKTI).62 In the epidermis, loss of function of LEKTI results in hyperactivation of serine proteases and peeling of the cornified layer. However, precise pathomechanisms for the hair symptoms remain unknown.
KID syndrome shows either autosomal dominant or recessive inheritance patterns. The autosomal dominant form of the disease is known to be caused by pathogenic variants in the GJB2 gene encoding a GAP junction protein, connexin 26.63 Recently, it has been reported that the autosomal recessive form results from pathogenic variants in the AP1B1 gene encoding an adaptin protein.64 It is suggested that AP1B1 deficiency results in a specific defect in clathrin-coated vesicles within the cytoplasm, which is most likely the main mechanism for the pathogenesis of KID syndrome.
Most cases of IFAP syndrome show an X-linked recessive inheritance pattern, although some families show autosomal dominant inheritance. The X-linked recessive form is caused by pathogenic variants in the MBTPS2 gene encoding an enzyme named site-2 protease (S2P),65 and the autosomal dominant form occurs through pathogenic variants in the SREBF1 gene encoding sterol regulatory element-binding protein 1 (SREBP-1).66 Notably, S2P digests SREBP-1 at the Golgi apparatus, after which the N-terminus of SREBP-1 moves into the nucleus and regulates expression of many genes (Fig. 3). Therefore, the two causative genes for IFAP syndrome are closely related.
At present, topical minoxidil is widely used for several hereditary hair diseases including ARWH,67 because it can stimulate hair matrix cells and promote hair growth. However, the effect is limited, and new treatments need to be developed based on the pathomechanisms of each disease. For example, in patients with ARWH caused by LIPH gene variants, it is believed that the amount of LPA is markedly reduced in the HFs. Therefore, topical application of LPA or its analogues can theoretically improve hair symptoms.
Notably, significant progress has been made toward establishing a therapy for HED caused by EDA gene variants. It is expected that development of the symptoms can be prevented by supplementation of EDA-A1 at the correct stage of the fetal period. Indeed, when recombinant EDA-A1 fused with the Fc domain of immunoglobulin G1 (Fc-EDA-A1) was injected into the amniotic cavity at gestational weeks 26 and 31, the symptoms of HED of the newborn were significantly improved.68 In particular, both the number and the function of eccrine sweat glands were dramatically rescued.68 However, for complete prevention of the disease, Fc-EDA-A1 should be supplemented at an earlier gestation stage.
It has recently been reported that dupilumab was effective for Netherton syndrome.69,70 This result suggests that the strategy of drug repositioning should also be considered for other hereditary diseases in the future.
There remain many hereditary diseases for which the molecular basis is still undefined. Identification and characterization of causative genes for hereditary hair diseases, as well as elucidation of functional relationships between these genes, can contribute to reveal complex mechanisms for morphogenesis and development of HFs in humans.
All hereditary hair diseases are intractable and can severely affect quality of life of the affected individuals. To date, none of the non-syndromic forms have been approved as a designated intractable disease in Japan. Nevertheless, our efforts in the Project for Research on Intractable Diseases by the Ministry of Health, Labour, and Welfare of Japan have enabled us to obtain certain achievements. For example, several syndromic forms, such as Netherton, KID, and IFAP syndromes, have been recognized as designated intractable diseases. In addition, HED has recently been approved as a specific pediatric chronic disease. However, because the condition is not accepted as a designated intractable disease, adult patients do not qualify for government support.
In future studies, investigations of hereditary hair diseases should continue in wet labs to further elucidate the contributing molecular factors. Such advances will see the emergence of further treatment options and the recognition of more hereditary hair diseases as designated intractable diseases.
This study was supported in part by a Grant-in-Aid for Scientific Research (C) (KAKENHI 21K08347) from the Japan Society for the Promotion of Science to Y.S. and by the “Research on Measures for Intractable Diseases Project” through matching fund subsidies (20FC1052 and 22FC1015) from the Ministry of Health, Labour, and Welfare of Japan.
The author declares that no conflict of interest exists.