Technical Report
Immuno-Mass Spectrometry Workflow for Quantification of Serum α-Fetoprotein Using Antibody-Immobilized Magnetic Beads and Modified Eluents
2023 年 12 巻 1 号 p. A0122

詳細
2023 年 12 巻 1 号 p. A0122
Immuno-mass spectrometry (MS) is a powerful method for the quantitative analysis of low-abundance proteins in biological specimens. In these procedures, collecting specifically and efficiently the target protein antigens from the antigen–antibody complex generated on the surface of nanocarrier beads is crucial and can be performed by hydrolyzing the proteins directly on the beads or after elution. Herein, we optimized the conditions of the immunoaffinity purification via elution using serum α-fetoprotein (AFP) as a model and its specific antibody immobilized covalently on magnetic beads. Antibody-coated beads were incubated with human serum spiked with standard AFP for antigen–antibody reaction. AFP was then eluted from the beads using various eluents, including organic solvents, to optimize the elution conditions. After proteolytically hydrolyzing the eluted protein, stable isotope-labeled standard peptides were added to the hydrolysate to quantify the eluted AFP via liquid chromatography–tandem MS. Using an optimized workflow for quantitative analysis afforded a correlation between the amount of spiked AFP and heavy to light ratios calculated based on peptide ion peak areas, from which an endogenous AFP concentration of 2.3±0.6 ng/mL was determined in normal serum; this is consistent with previous reports using radioimmunoassay methods. The present immuno-MS workflow could apply to the detection and quantitation of other low-abundance biofluid biomarkers.
The quantitative analysis of peptides hydrolyzed from biomarker proteins using liquid chromatography–tandem mass spectrometry (LC–MS/MS) and stable-isotope-labeled (SIL) peptide internal standard dilution has emerged as a promising clinical tool. Its excellent selectivity and high precision1–3) allow using signature peptides as stoichiometric surrogates for absolute protein quantitation after treatment with specific protease(s). However, to assess trace amounts of proteins in complex samples such as serum, the target protein and peptides must be purified before the MS analysis.4)
Immuno-MS peptide measurement combined with immunoaffinity purification of proteins is a powerful method for the detection of low-abundance proteins in biological specimens. Several research groups have demonstrated that the purification of target proteins or peptides using antibody-immobilized magnetic particles (beads) before MS analysis contributes to achieving high sensitivity and reproducibility.5–10) Magnetic beads are convenient tools used in a wide range of molecular biology processes, such as purification of nucleic acids, separation of proteins, and detection of biomarkers.11–15) In addition, they can be adapted to automation of separation processes due to their magnetism.
In immuno-MS, two types of procedures are frequently used to collect fragment peptides of an antigen from an antibody–antigen complex: direct hydrolysis by protease(s) on the complex coated on-beads (on-beads hydrolysis) and hydrolysis after elution of the antigen from the complex. Moreover, immuno-capturing of both proteins and peptides has been reported to achieve relatively high sensitivity.16,17) In this method, the initial step is the immuno-capturing of the antigen, followed by its elution with an aqueous acid solution. After tryptic hydrolysis, the surrogate peptide is extracted by an antipeptide antibody and quantified by LC–MS/MS.17–19)
In these immuno-MS procedures, the recovery of proteins and peptides from the antibody–antigen complex is a crucial step. Levernæs et al. studied the convenience of eluting the proteins before hydrolysis for the immune-MS detection methods by comparing them with the on-beads hydrolysis.10) However, determining whether on-bead hydrolysis and the elution method were suitable for immuno-MS was difficult because both approaches depended on the properties of the surrogate antigen or antibody peptides.20) Nevertheless, we considered that conducting hydrolysis after elution also reduces the contamination of proteolytic peptides generated from antibodies on magnetic beads, thereby reducing the load on LC–MS/MS columns and MS system contamination and extending the lifetime of stable continuous measurements. Moreover, expensive antibodies could be recycled in some cases.
In this study, we examined the optimal conditions to elute undigested antigens from the antigen–antibody complex generated on the surface of magnetic carrier beads using α-fetoprotein (AFP), a diagnostic marker for hepatocellular carcinoma, as an experimental model in spiked or nonspiked normal serum and compared the results with those obtained by conducting on-beads hydrolysis (Fig. 1). By demonstrating the quantification of AFP, we revealed AFP concentrations below 10 ng/mL in normal serum.21) The concentration range of serum proteins is extremely wide, which often impairs the detection of candidate biomarker proteins, including AFP, in conventional whole serum shot-gun approaches, where they are present in minute amounts.22)
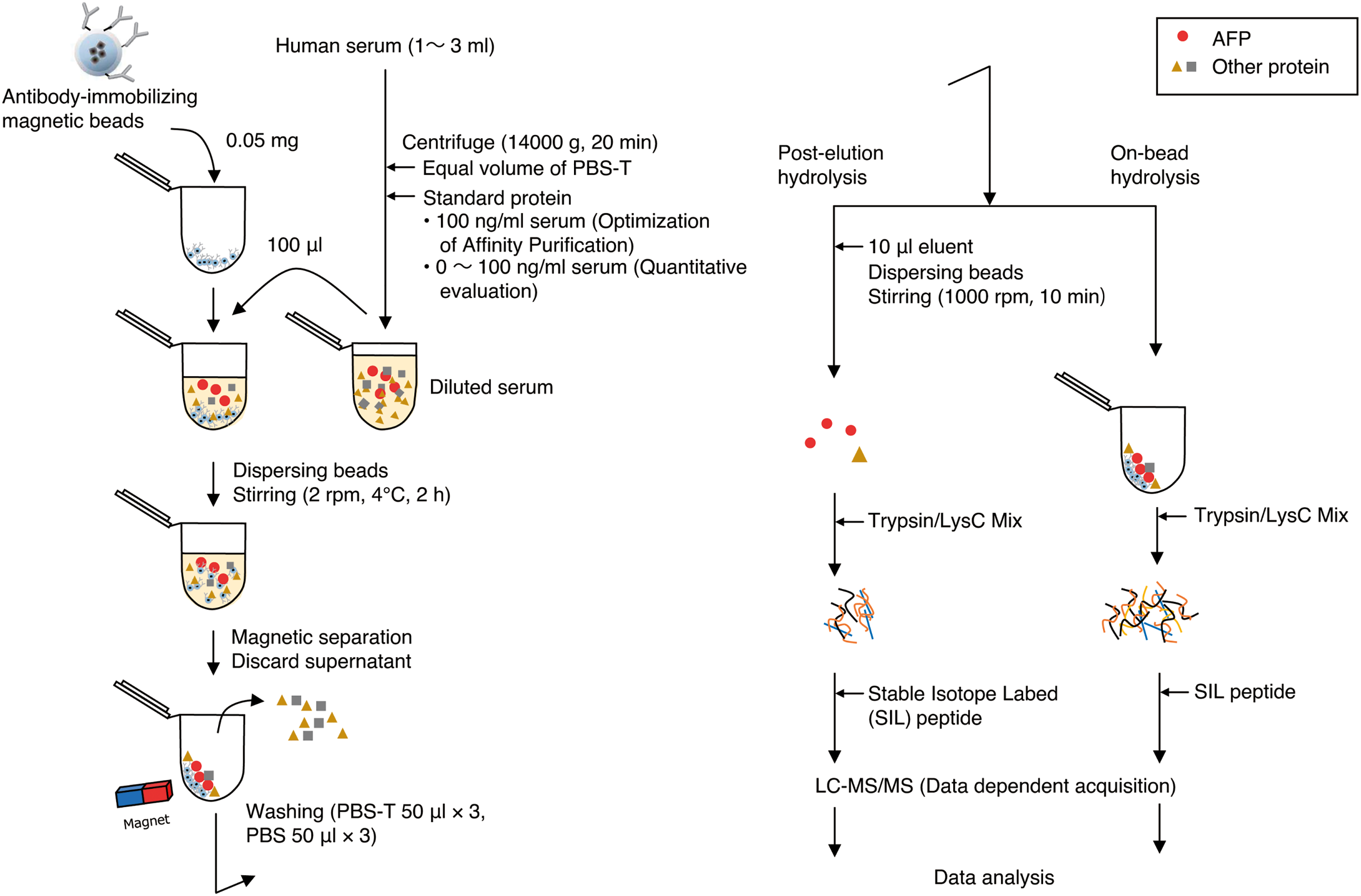
The three SIL peptides (TFQAITVTK, YIQESQALAK, and NIFLASFVHEYSR) for AFP quantitation (more details are provided in the Results and Discussion section) were synthesized at JPT Peptide Technologies (SpikeTides TQL, Berlin, Germany). These peptides were labeled with stable isotopes at the C-terminal Arg (13C6; 15N4, +10.008 Da) or Lys (13C6; 15N2, +8.014 Da), and they contained a sequence tag (JPT quant-tag; JPT Peptide Technologies) used for accurate peptide quantification. The sequence tags were cleaved by trypsin following the manufacturer’s protocol to obtain the final peptides. The peak intensity ratios of these SIL peptides and their corresponding native forms from AFP produced by tryptic hydrolysis were used in the quantitative calculation.
2.2. Preparation of AFP-spiked serum samplesNormal human serum was purchased from Sigma–Aldrich Co. (St. Louis, MO, USA) and stored at −80°C before analysis. The frozen serum was completely thawed and diluted with an equal volume of phosphate-buffered saline (PBS; 1.06 mM KH2PO4, 2.97 mM Na2HPO4, and 155 mM NaCl; and pH 7.4) containing 0.05% (v/v) Tween-20 (PBS-T). The diluted serum sample was centrifuged at 14,000×g for 20 min at 4°C. For the optimization of the elution conditions, an aliquot of serum sample was spiked with standard AFP to a final concentration of 20 ng/mL, except for the analysis of endogenous AFP. For the quantitative analyses, AFP was spiked in six serum samples at different concentrations (0, 2, 10, 20, 50, and 100 ng/mL), except for the analysis of endogenous AFP.
2.3. Coupling of the antibody and magnetic beadsThe covalent coupling of N-hydroxysulfosuccinimide (NHS) FG beads (Code TAS8848N1141; Tamagawa Seiki Co., Nagano, Japan) and the anti-AFP antibody (clone 1D5; Mikuri Immunology Laboratory, Osaka, Japan) was performed according to the manufacturer’s protocol. In brief, NHS FG beads were treated with methanol, followed by incubation with the anti-AFP antibody solution at 4°C for 60 min. The magnetic beads were mixed with 1 M of 2-aminoethanol (pH 8.0) with 0.1% NP-40 at 4°C for 16–20 h to quench the conjugation reaction; washed five times with a washing buffer solution composed of 10 mM HEPES–NaOH, pH 7.9, 50 mM KCl, 1 mM EDTA, and 10% glycerol; and stored in the washing buffer at 4°C.
2.4. Affinity purification of AFP from human serum using the anti-AFP antibody-conjugated magnetic beadsThe antibody-conjugated beads were washed three times with PBS-T and suspended at a concentration of 5 mg/mL. Then, 10 μL of the suspension, containing 50 μg of beads, was added with 100 μL PBS-T in a 1.5-mL tube. After the tubes were shaken on a vortex mixer, spun down, and placed in a magnetic rack, the supernatant was removed. Then, 100 μL of the serum sample was added to the tube and mixed with the antibody-conjugated beads by gently shaking using a rotator (Model NRC-20D; Nissinrika Co., Tokyo, Japan) at 4°C for 2 h. The tubes were placed in the magnetic rack to collect the beads. While on the magnet, the supernatant was removed and discarded. The beads were rinsed with 50 μL of PBS-T and shaken on a vortex mixer for 2 s to spread the beads, and the supernatant was then discarded. This step was repeated two additional times, followed by three times rinsing with PBS. The rinsed beads were then suspended in 10 μL of elution buffer (e.g., 0.1% trifluoroacetic acid [TFA], 50% acetonitrile [ACN]), shaken on a vortex mixer for 2 s, and incubated and shaken at 1000 rpm for 10 min at 2°C in a ThermoMixer (Eppendorf SE, Hamburg, Germany) to elute the proteins. The composition of the eluent is listed in Table S1. Here, relatively radical elution conditions were selected with the goal of efficiently eluting the target molecule. For on-beads hydrolysis, the elution step by elution buffer was omitted. The tubes were placed in the magnetic rack. While on the magnet, the eluent was recovered and transferred into a new tube. The resultant eluent was dried in a vacuum evaporator. The affinity purification step was performed with three technical replicates.
2.5. Preparation of peptide mixturesThe eluted proteins or rinsed beads (for on-beads hydrolysis) suspended in 10 μL of buffer solution, consisting of 50 mM Tris–HCl, pH 8.0, and 8 M urea, were reduced by 10 mM dithiothreitol solution at 37°C for 20 min, followed by cysteine alkylation using 10 mM iodoacetamide for 20 min in the dark. The resultant samples were diluted eight-fold to a final concentration of 1 M urea, 50 mM Tris–HCl, and pH 8.0, and then subjected to protein hydrolysis by adding 4 μL of a 100-μg/mL Trypsin/Lys-C Mix solution (Promega Co., Madison, WI, USA) at 37°C for 16 h. To stop the hydrolysis, TFA was added into protein hydrolysates to a final concentration of 1%. The acidified hydrolysates were loaded onto Stop and go extraction (STAGE) tips23) filled with C18 and poly(styrene–divinylbenzene) (SDB) copolymer Empore disk membranes (3M Co., St. Paul, MN, USA) equilibrated with 0.1% TFA. The C18 and SDB materials were washed with 0.1% TFA in 2% ACN followed by a serial elution of peptides with 30% and 60% ACN in 0.1% TFA. All peptide fractions were collected together and lyophilized.
2.6. LC–MS/MS analysisThe peptide mixture was analyzed using an LC–MS/MS system with a Q Exactive Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) in a fully automated manner. Briefly, peptide separation was performed using an UltiMate 3000 RSLCnano liquid chromatograph (Thermo Fisher Scientific) containing a C18 capillary LC column needle (Nano HPLC capillary column, 75-μm i.d., 150-mm length, 3-μm particle size; Nikkyo Technos Co., Tokyo, Japan). The mobile phase consisted of formic acid, ACN, and water at a volume ratio of 0.1 : 0 : 100 for mobile phase A and 0.1 : 90 : 10 for mobile phase B. The peptide solution was dissolved in water/ACN/TFA solution (98 : 2 : 0.1 v/v/v) and loaded on a precolumn (Acclaim PepMap μ-Precolumn, 300-μm i.d., 5.0-mm length; Thermo Fisher Scientific) equilibrated with mobile phase A. The peptides concentrated and purified on the precolumn were injected onto the capillary column by valve switching. The peptides were continuously eluted at a rate of 350 nL/min in a gradient mode as follows: An initial ratio of 5% of mobile phase B was increased linearly to 40% B during 40 min, followed by an increase to 95% B during the next 10 min. After washing with a nongradient flow at 95% B for 10 min, the column was equilibrated again with 2% B for the next separation. The total elution time was 60 min. The LC effluent from the column needle was directly interfaced with an electrospray ion source on the mass spectrometer. Protonated peptides in the gas phase were analyzed sequentially by MS/MS in a positive ion mode with a full-range scan in an m/z range of 300–1500 and subsequent product ion scans for each of the ten most intense ions in the full-scan mass spectrum. The full-range scan settings included a mass resolution of 30,000 (full width at half maximum [FWHM]); autogain control (AGC) of 5×105; and a lock mass of m/z 391.2843 and 445.1200. The product ion scan was performed under conditions of a mass resolution of 17,500 (FWHM), the maximum ion injection time of 60 ms, AGC of 1×105, an intensity threshold of 5000, 27% normalized collision energy, 1.6 Da isolation m/z width, and dynamic exclusion for 15 s.
2.7. LC–MS/MS data processing and sample quantificationTo identify the peptide or protein, the MS/MS data were extracted using the Progenesis QI for proteomics software (version 2.0; Nonlinear Dynamics Ltd., Newcastle, UK), and the MS/MS spectra were screened for the amino acid sequence of AFP using the Mascot (Matrix Science, Boston, MA, USA) algorithm (version 2.5). The parameters used for the screening were peptide mass tolerance of ±5 ppm, MS/MS tolerance of ±0.02 Da, fixed modifications of carbamidomethylation (Cys, +57.021), and variable modifications of oxidation (Met, +15.995). For the quantification analysis, LC–MS/MS raw files were processed using the Skyline software (version 3.4; MacCoss Laboratory, University of Washington, WA, USA) for the generation of the extracted-ion chromatograms and peak integration. The quantitation was performed using the peak areas of the top three resulting precursor ions (i.e., M, M+1, and M+2) corresponding to the SIL heavy and native light m/z value of each peptide. The isotopic envelope (M, M1, M2) extracted from the MS1 scan by MS1 filtering was visually confirmed to be consistent with the theoretical isotopic distribution, allowing peak picking adjustments to be made accordingly.24)
2.8. Data analysisAll subsequent data analyses were performed in Microsoft Excel to generate standard deviations and calibration curves. Standard deviations ware calculated using triplicate analyses for each known concentration. Calibration curves were generated by plotting the peak area ratios of native peptide and SIL peptide vs. the concentrations of AFP-spiked serum. The limit of detection (LOD) and limit of quantitation (LOQ) were calculated using the following formulas: LOD=3Sa/b and LOQ=10Sa/b, where Sa is the standard deviation of the minimum detectable concentration with a relative standard deviation ≤20% and b is the slope of the standard curve.2)
2.9. Selection of the quantitative peptidesFirst, the affinity-purified AFP was analyzed using a label-free quantification method. Affinity-purified AFP was collected using magnetic beads with immobilized antibodies from nonspiked serum. After elution and hydrolysis with a Trypsin/Lys-C Mix solution, the resulting peptides were subjected to LC–MS/MS analysis. LC–MS/MS raw data were then processed using Progenesis QI to generate peak lists, which were searched against the Swiss-Prot database using the Mascot software. The identifications were filtered to satisfy false discovery rate (FDR) of 1% on peptide. Eighteen unique AFP peptides with a Mascot score of >30 were considered to be positively identified peptides (Fig. S1), among which three candidate peptides were selected for quantification according to the following criteria: (1) no missed cleavage site for trypsin, (2) no Glu residue at the N-terminus and neither Cys nor Met residue in the amino acid sequence for chemical stability, and (3) relatively high mass response. The corresponding SIL peptides were synthesized, dissolved in a water/ACN/TFA solution (98 : 2 : 0.1 v/v/v), and analyzed by LC–MS/MS. Of these three SIL peptides in the solution, no peak was detected for SIL peptide NIFLASFVHEYSR (data not shown). The other two SIL peptides were detected with sufficient intensity for quantitative analysis. The linear regression analysis of the signals of the two internal standard peptides in the solution resulted in the determination of the regression coefficients of 0.9997 and 0.995 for the SIL forms of TFQAITVTK and YIQESQALAK, respectively, demonstrating sufficient quantitative linearity (Fig. S2). Consequently, these two peptides were used as a signature peptide for the following quantitative analysis.
The present results show that performing an optimized elution with organic solvents before the protein hydrolysis facilitates the recovery of the antigenic proteins from the antibody immobilized on magnetic beads. Our method was evaluated by conducting an MS-based quantitative analysis of AFP-spiked sera. Herein, antibody-immobilized beads with NHS were used in the immunoaffinity method. The most considerable advantage of this scheme is the reduction of the elution of antibodies using beads to which only the antibodies are directly covalently attached. Moreover, the beads can be stored for long periods, with the antibodies still bound to them. However, this scheme requires caution because by depending on the orientation in which the antibodies bind, the activity of the bound antibodies may be reduced.
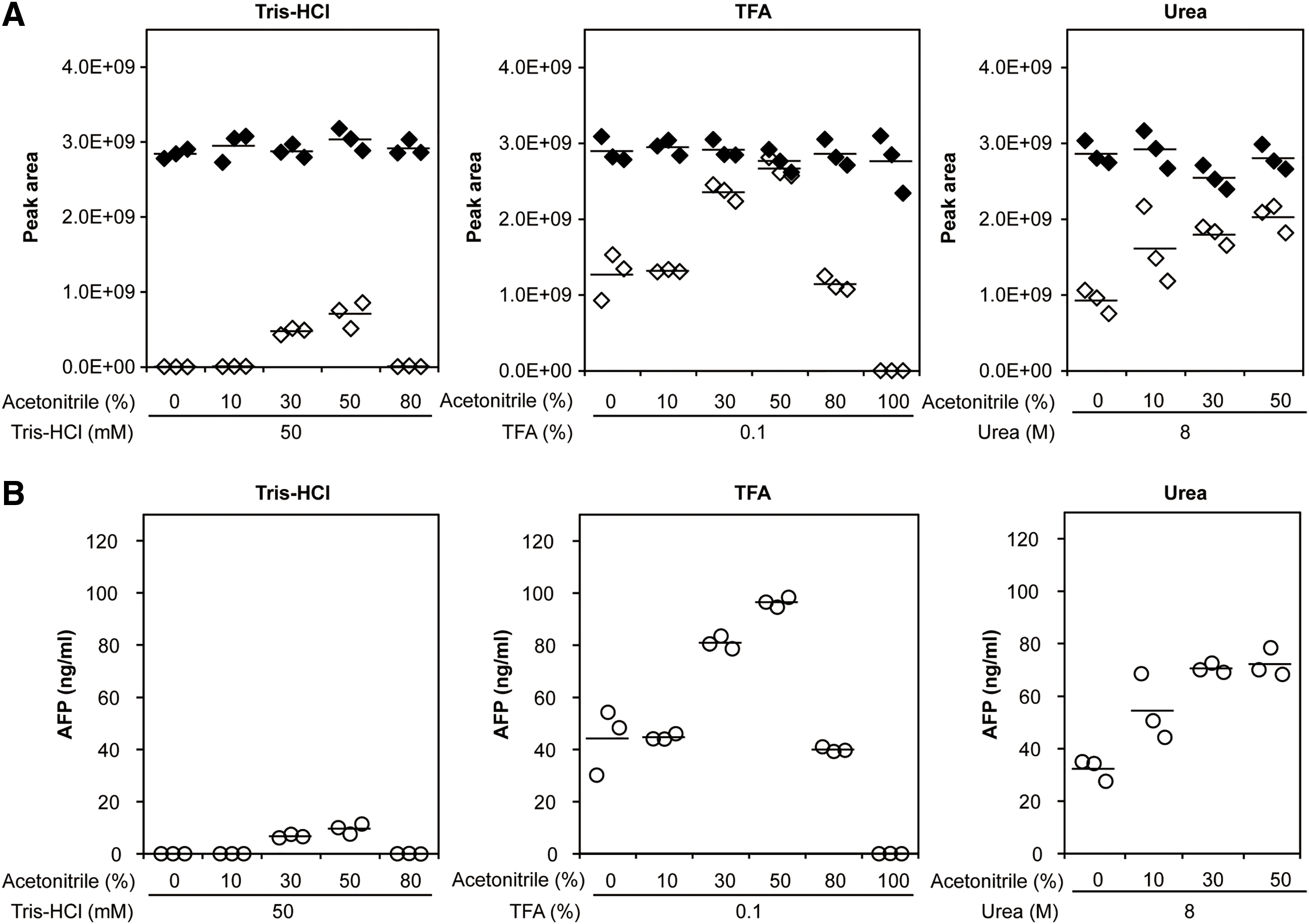
3.1. Comparison between postelution hydrolysis and on-beads hydrolysisThe collection of trypsinized peptides from antibody-coated magnetic beads was performed following two distinct proteolytic hydrolysis procedures, i.e., hydrolysis after elution (postelution hydrolysis) and on-beads hydrolysis. These two methods were quantitatively compared. The following three eluates were used to elute the antigen from the antigen–antibody complex: (1) 50 mM Tris–HCl, pH 8.0 (Tris), (2) 8 M urea, 50 mM Tris-HCl, pH 8.0 (Urea), and (3) 0.1% TFA (TFA). The use of 8 M urea, which denatures proteins, promotes the release of the target proteins from the antibody; 0.1% TFA was used as a common acidic enhancer to elute the antigen from the antibody; and Tris was used as a negative control because it is not seemed to affect the elution from an antibody. The peak intensities obtained after on-beads hydrolysis were higher for both the native forms of TFQAITVTK and YIQESQALAK from AFP but lower for the SIL forms compared with the postelution methods (Fig. 2). On the on-bead hydrolysis, the ratio of the peak intensity of the SIL peptides and the AFP-derived native peptides was 0.92. The amount of spiked SIL peptides (75 fmol) corresponded to the AFP added to the serum (100 ng/mL), thus indicating a high recovery rate. The conversion to the mass concentration (ng/mL) was performed by multiplying the molecular mass of the intact AFP (67.3 kDa).

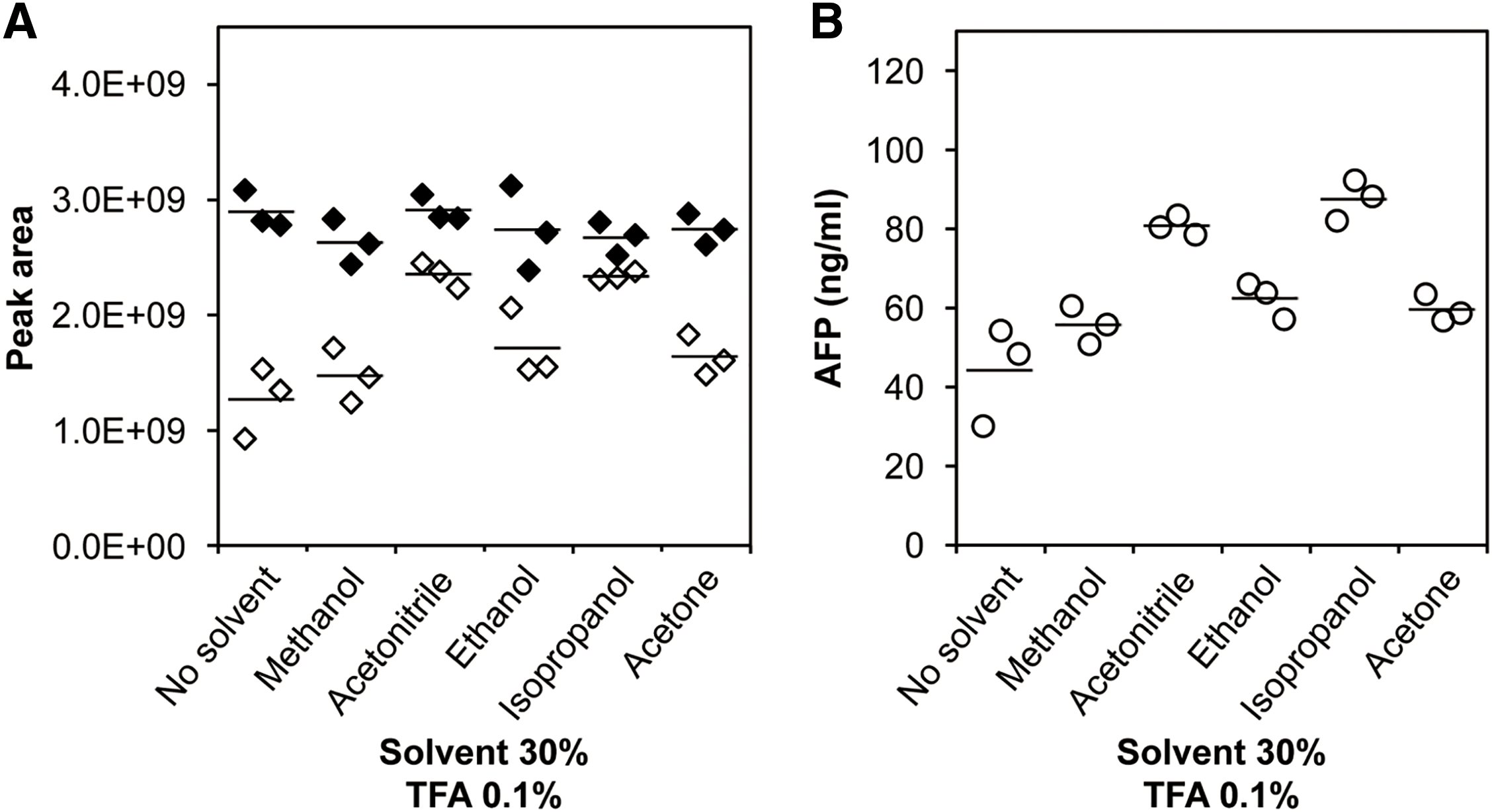
To increase the elution amount, the effect of using a solution containing 0.1% TFA and 30% organic solvent on the dissociation of the antigen from the antibody was investigated. Ethanol, methanol, ACN, isopropanol, and acetone were used as the organic solvent. The addition of any organic solvent resulted in an increase in the recovery of the signature peptide of TFQAITVTK compared with that obtained without any additive (Fig. 3). When ACN and isopropanol were added as the eluent, the recovery amount of signature peptides was increased by 1.8 and 2.0 times, respectively, compared with that in the absence of organic solvents. A similar trend was observed for YIQESQALAK (Fig. S3).
Next, for the dissociation of the antigen from the antibody, the optimum concentration of ACN was investigated. Between isopropanol and ACN, which were highly effective in elution, ACN was selected for optimization because there is no clear difference in the elution effect between them, and ACN is a suitable solvent often used for proteomics pretreatment due to its low chemical reactivity, high miscibility with water, low viscosity, and low ultraviolet cutoff rate. The six compositions of TFA-based eluents containing ACN, i.e., 0.1% TFA and 0% ACN (0% ACN), 0.1% TFA and 10% ACN (10% ACN), 0.1% TFA and 30% ACN (30% ACN), 0.1% TFA and 50% ACN (50% ACN), 0.1% TFA and 80% ACN (80% ACN), and 0.1% TFA and 99.9% ACN (100% ACN), were compared. The amount of the signature peptide eluted from the antigen–antibody complex reached a maximum using the 50% ACN eluent (Figs. 4 and S4). The amount of elution decreased with 80% ACN and 100% ACN, and 100% ACN was hardly eluted. This could be due to a decrease in the antigen solubility in the eluent at ACN concentrations above 50%. The detection intensity of nonspecific protein-derived peptides, except AFP, increased under the elution condition of 0.1% TFA and 50% ACN compared to that for 0.1% TFA without ACN (data not shown). Meanwhile, for the 8 M urea-based eluents, the amount of signature peptide eluted with 40% ACN was larger than that eluted with 0% ACN, 10% ACN, and 30% ACN. However, when the ACN concentration exceeded 40%, urea was not completely dissolved. Therefore, an ACN concentration of 40% or more in 8 M urea was not suitable for the elution. Using Tris-HCl-based eluents, the signature peptide was slightly recovered with 30% ACN and 50% ACN. These results indicate that a combination of ACN and TFA or urea is required to improve the dissolution effect. Moreover, optimization of these compositions for cleaning contaminants might enhance the purity of AFP. Furthermore, the 50% ACN TFA-based eluent showed the highest recovery rate, which was similar to that obtained via on-beads hydrolysis, and the signal intensity of signature peptides was higher than that in the on-beads hydrolysis method. The background peak, which was smaller using the 0.1% TFA and 50% ACN eluent than using the on-beads method, might attenuate the reduction in the peak intensity by the ionization suppression effect. Ionization matrix effects refer to the suppression or enhancement of ionization by coeluted extracted substances in the biological matrix.25) Ionization suppression caused by high abundance peptides may influence the detectability of low-abundance peptides using MS. Levernæs et al. reported a lower signature peptide recovery rate for elution with 0.1% TFA and 2% formic acid than for on-beads hydrolysis, which is in accord with our results.10) As mentioned above, 50% ACN in 0.1% TFA afforded a high recovery rate of signature peptides similar to that of the on-beads method, an indication of the effectiveness of our method. Furthermore, this elution method allowed us to recover almost all AFP from the antibody beads. In other words, when using the 0.1% TFA and 50% ACN eluent, the quantitative values were 96.4 ng/mL for the TFQAITVTK peptide and 112.0 ng/mL for the YIQESQALAK peptide. These values correspond to 94% and 110% of the added 100 ng/mL AFP protein, respectively, even after subtracting 2.3–2.4 ng/mL endogenous AFP (see below). However, the elution effect of ACN may be influenced by antigen solubility in the eluent and nonspecific binding forces (e.g., hydrophobic interactions) between the antibody, antigen, and magnetic beads except for the covalent bond between the antibody and magnetic beads. These results warrant further investigation. Moreover, adding organic solvents into the immunoprecipitation eluent risks precipitating proteins (especially, high molecular-weight proteins). Notably, this method, which involves using the 50%-ACN TFA-based eluent, may cause precipitation when used for proteins other than AFP.
The linearity of the quantitative method using eluents comprising 0.1% TFA and 50% ACN was investigated. For TFQAITVTK, a linearity of >0.9964 was obtained in the spiked-AFP concentration range 0–100 ng/mL (0, 2, 5, 10, 20, 50, and 100 ng/mL). Similarly, the linearity for YIQESQALAK was >0.9966 at the same concentration range (Fig. 5). Using the standard curve regression equations shown in Fig. 5, the concentration of endogenous AFP was calculated to be 2.3 ng/mL for TFQAITVTK and 2.4 ng/mL for YIQESQALAK, which is in agreement with previous reports on AFP concentrations in healthy human serum (<10 ng/mL).14,26) The LOD of TFQAITVTK and YIQESQALAK was 0.25 and 0.45 ng/mL, respectively, and the LOQ of TFQAITVTK and YIQESQALAK was 0.84 and 1.50 ng/mL, respectively.

The parallel reaction monitoring and multiple reaction monitoring methods, which are generally advantageous in terms of sensitivity and accuracy in quantitative analysis, were not employed in this study. Quantification was performed by MS1 data in the data-dependent acquisition (DDA) mode, which is easily set up and has the advantage that proteins other than the target protein can be observed simultaneously. Herein, we found that efficient purification and elution by the immunoaffinity method provided sufficient sensitivity over the concentration range of AFP in normal serum even in the DDA mode.
Peptides were recovered with high efficiency by performing the hydrolysis after eluting the antigen, indicating that the intact protein can be recovered with a high yield before hydrolysis. After further optimization, this method could facilitate the analysis of small amounts of serum proteins in their intact forms. Furthermore, antibodies were kept intact and not exposed to proteolytic hydrolysis in the proposed method. If we can find mild conditions for recovery of antigen that do not cause the decrease or loss of antibody activity, we can reuse the antibodies. Alternatively, our method elutes antigens efficiently, can be specifically detected via MS, and would allow the use of relatively inexpensive and poorly performing polyclonal antibodies. Herein, we employed magnetic beads, which are suitable for automation. In the future, it will be possible to fully or semi-automate the system by combining it with an automated pipetting machine or a device that can manipulate magnetic beads by the permanent magnets and process many samples simultaneously.
In this study, we optimized an elution method to effectively elute AFP from antibody-immobilized magnetic beads before hydrolysis. The recovery rate of AFP signature peptide via hydrolysis after elution using an eluent comprising 0.1% TFA and 50% ACN was increased 1.8 times over that of elution experiments performed using eluents containing 0.1% TFA and without ACN, with the signal intensity of the signature peptide being higher than that achieved using on-beads hydrolysis methods. The linearity of our quantitative method using eluents was >0.996 in the spiked-AFP concentration range of 0–100 ng/mL. The concentration of endogenous AFP in normal human serum, which has a low concentration of AFP (<10 ng/mL), was successfully calculated to be 2.3–2.4 ng/mL using a standard curve regression equation, outperforming conventional proteomics, which is difficult for the detection of low-AFP concentrations in normal serum. Although further studies with several antibodies and antigens are needed for its validation, this method could facilitate diagnosis usage of small amounts of protein in serum and the detection and quantitation of other low-abundance biofluid biomarkers.
The authors would like to thank the staff at Proteome Research Center in Yokohama City University’s Advanced Medical Research Center for their advice and support.
Mass Spectrom (Tokyo) 2023; 12(1): A0122
Table S1 Eluents used for the elution of AFP bound to antibody-coated magnetic beads. AFP, α-fetoprotein.
Fig. S1 Sequence map of AFP. AFP, α-fetoprotein.
Fig. S2 Calibration curves of peak area vs. SIL peptide injected on the column. SIL, stable isotope labeled.
Fig. S3 Effects of using organic solvents for eluting AFP from antibody-coated magnetic beads (data from the YIQESQALAK peptide). AFP, α-fetoprotein.
Fig. S4 Optimization of ACN concentrations in the elution solution (data from the YIQESQALAK peptide). ACN, acetonitrile.