Review
25 Years Responding to Respiratory and Other Viruses with Mass Spectrometry
2023 年 12 巻 1 号 p. A0136

詳細
2023 年 12 巻 1 号 p. A0136
This review article presents the development and application of mass spectrometry (MS) approaches, developed in the author’s laboratory over the past 25 years, to detect; characterise, type and subtype; and distinguish major variants and subvariants of respiratory viruses such as influenza and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). All features make use of matrix-assisted laser desorption ionisation (MALDI) mass maps, recorded for individual viral proteins or whole virus digests. A MALDI-based immunoassay in which antibody–peptide complexes were preserved on conventional MALDI targets without their immobilisation led to an approach that enabled their indirect detection. The site of binding, and thus the molecular antigenicity of viruses, could be determined. The same approach was employed to study antivirals bound to their target viral protein, the nature of the binding residues, and relative binding affinities. The benefits of high-resolution MS were exploited to detect sequence-conserved signature peptides of unique mass within whole virus and single protein digests. These enabled viruses to be typed, subtyped, their lineage determined, and variants and subvariants to be distinguished. Their detection using selected ion monitoring improved analytical sensitivity limits to aid the identification of viruses in clinical specimens. The same high-resolution mass map data, for a wide range of viral strains, were input into a purpose-built algorithm (MassTree) in order to both chart and interrogate viral evolution. Without the need for gene or protein sequences, or any sequence alignment, this phylonumerics approach also determines and displays single-point mutations associated with viral protein evolution in a single-tree building step.
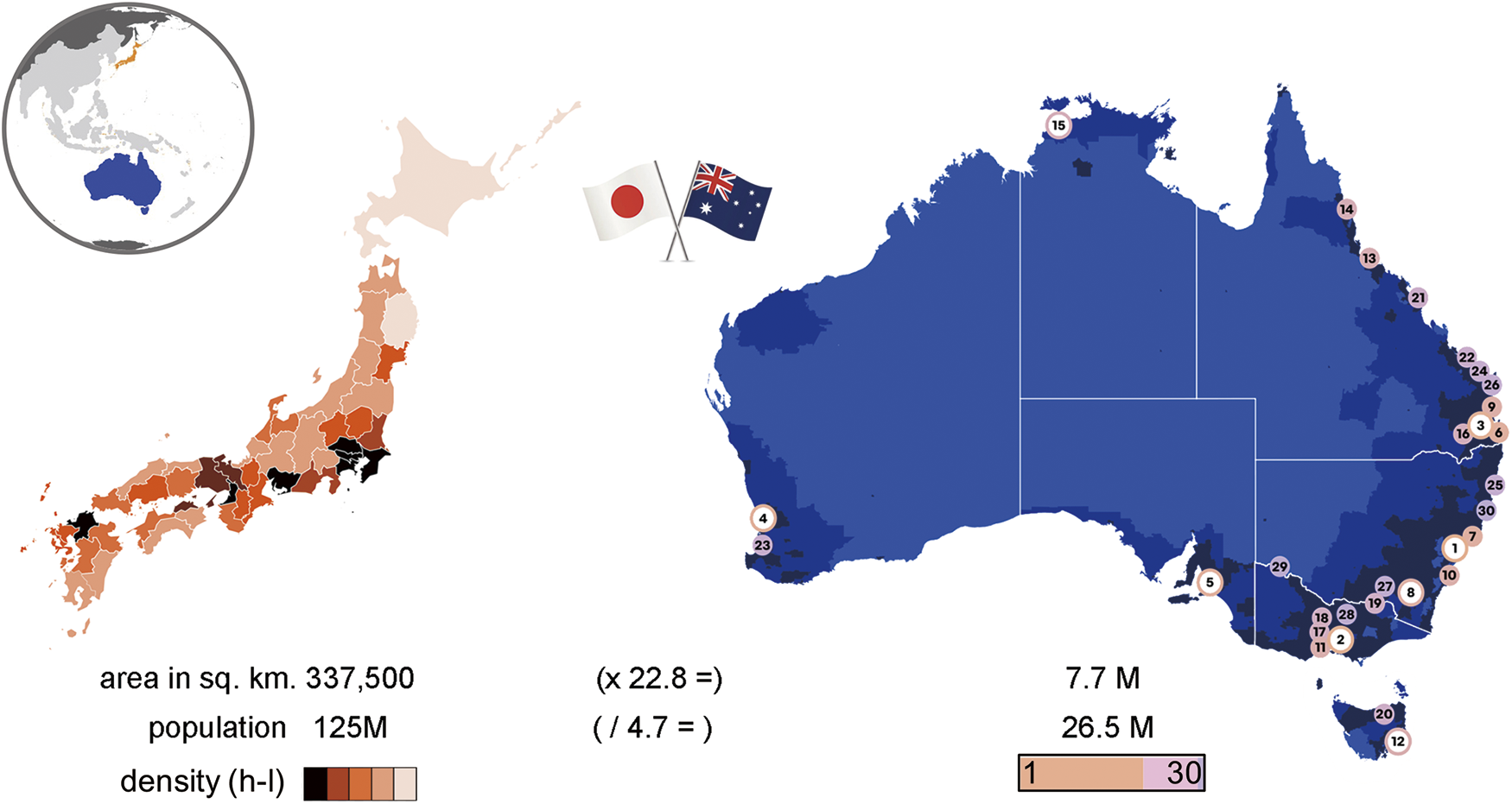
Respiratory viruses have had a significant impact on human health over the centuries, as exemplified by the recent severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) (COVID-19) pandemic.1) Easily transmissible among humans and animals, such viruses evolve in a relatively haphazard manner where the most transmissible and virulent strains survive.2) Their rapid mutation rates enable them to evade natural immune responses and quickly spread throughout a population.3) Human respiratory viruses cause a spectrum of symptoms and disease, contributing to significant morbidity, mortality, and economic losses.2,4) This is particularly pronounced in high-density and large population centres as found in much of eastern Japan and coastal sections of eastern Australia.
The Japanese island of Honshu accommodates 80% of the population comprising over 100 million individuals, many concentrated in some of Japan’s largest cities of Tokyo, Yokohama, Osaka, Nagoya, and Kyoto.5) Australia is a large island continent spanning 7.7 square kilometres, but most (near 90%) of its 26.5 million population reside within 50 km of its coastline.6) Most of these live along the eastern perimeter in Australia’s largest cities of Sydney, Melbourne, Brisbane, which account for half of the total population alone (Fig. 1). Greater Sydney has a population density of over 2000 people per square kilometre, over 500 times that estimated based on a national, total land area basis.
Respiratory viruses are transmitted among the population via four major modes of transmission2): direct physical contact, indirect contact with contaminated objects, or exposure to airborne virus-containing droplets and aerosols (Fig. 2). Little is known about the relative contribution of each mode to the total transmission of a particular virus, but high population densities are known to exacerbate transmission rates.

Prior to the development and deployment of any treatment regime, there is a need to rapidly detect viruses in the population and combine such evidence with epidemiological data,7,8) in order to manage and plan effective response strategies.
The detection and analysis of virus particles collected in human clinical specimens, swabs of surfaces, or air sampling provides the first frontline defence against a virus.9) A combination of non-molecular and molecular approaches can be employed,10) each aimed at determining some aspect of the virus’ character and transmissibility.
Currently, nucleic acid (DNA or RNA) detection and immunoassay methods are among the most popular means to quickly identify viral infection at the molecular level, regardless of the source.11–13) Polymerase chain reaction (PCR) methods remain the “gold standard” for virus detection, particularly following the development of diagnostic assays employing real-time quantitative PCR (RT-qPCR).14) Here fluorescently labelled DNA probes bind to target amplicons specific to a particular virus. The level of fluorescence measured is directly related to the initial concentration of virus in the sample (Fig. 2). While affording high specificity, an assay that produces quantitative results can take many hours and sample preparation is a delaying factor. Commercially available kits facilitate the isolation of viral DNA or RNA,15) although they add an associated cost to analysis that, together with the initial cost of the qPCR equipment itself, can impact their use and deployment in remote geographical regions and developing countries.
Immunoassays employ antibodies to detect viruses within a sample.12,13) Monoclonal antibodies, and certain recombinant forms of antibodies, provide singular-epitope specificity that is valued in diagnostics for the targeted detection of virus proteins. In virus identification, this is useful for the differentiation of viral types and subtypes. Immunoassays, such as the enzyme-linked immunosorbent assay (ELISA), are employed to measure an antibody response against a given antigen (Fig. 2). Both direct and indirect ELISA assays can confirm whether patients have antivirus antibodies in their sera following an immune response to viral infection.
Mass spectrometry (MS) represents an alternative to PCR-based approaches for the sensitive detection of virus levels in clinical and cell-cultured specimens.16) It offers the ability to detect and confidently assign microorganisms down to the mid-low 103 copy range.16) It also offers the ability to perform molecular-based immunoassays17) by treating viral proteins with monoclonal antibodies and monitoring that binding during or after release of bound protein with MS.
For over 25 years, my laboratory has shown that MS approaches can have benefits for such molecular analyses, and that they complement gene- and genome-based PCR methods. Since the late 1990s, my laboratory has developed and applied MS approaches for the study of viruses17) that has resulted in some 60 published works. Beginning with the application of a new matrix-assisted laser desorption ionisation (MALDI)-MS immunoassay to study viral antigenicity,17) without the need to immobilize either antibody or antigen, or isolate the immune complex, we applied the same technique to study the binding of antiviral drugs.18,19) We subsequently developed a high-resolution proteotyping approach to type, subtype, and establish the lineage20) and nature of subvariants,21) and used the same mass map data to chart and study the evolution of viruses (using “phylonumerics”),22–24) without the need for gene or protein sequences, or sequence alignments.
This review briefly highlights each of these approaches, which when pieced together (Fig. 3) form a coordinated MS platform to characterise viruses at the molecular level.

MALDI has been coupled to time-of-flight (ToF) mass analysers since the development of approach.25,26) Since then a large variety of compact commercial MALDI-ToF instruments have been produced, many deployed in laboratories dedicated to microbial identification27) and screening in clinical settings.28–30) Apart from their relative ease of operation by nonexperts, MALDI-based instruments offer advantages in terms of the ease of sample preparation, high sample throughput, short analysis times, and greater tolerance to salts than electrospray ionisation (ESI)-based approaches. ESI-LC-MS suffer from the often overlooked and underreported time to set up high pressure liquid chromatography (HPLC) conditions, and equilibrate and flush columns between runs. The use of reverse-phase columns in LC-MS experiments is also accompanied by some unavoidable sample loss to the column.
The incorporation of delayed ion extraction methods31,32) on MALDI-ToF instruments and the later coupling of MALDI sources to a wide range of scanning mass analysers33,34) have improved mass accuracy and resolution over single-pass ToF instruments. Kingdon or Orbitrap34) ion traps and ion cyclotron resonance (ICR) instruments35) confer the highest mass resolution, which improves confidence in virus identification and assignment. This article presents the application of MALDI-ToF, MALDI hybrid (quadrupole) Q-ToF, and high-resolution MALDI Fourier transform ion cyclotron resonance (FT-ICR) instruments in the study of respiratory viruses, given these have posed the greatest risk to human health over the past century,1) as exemplified by the recent SARS-CoV2 pandemic.
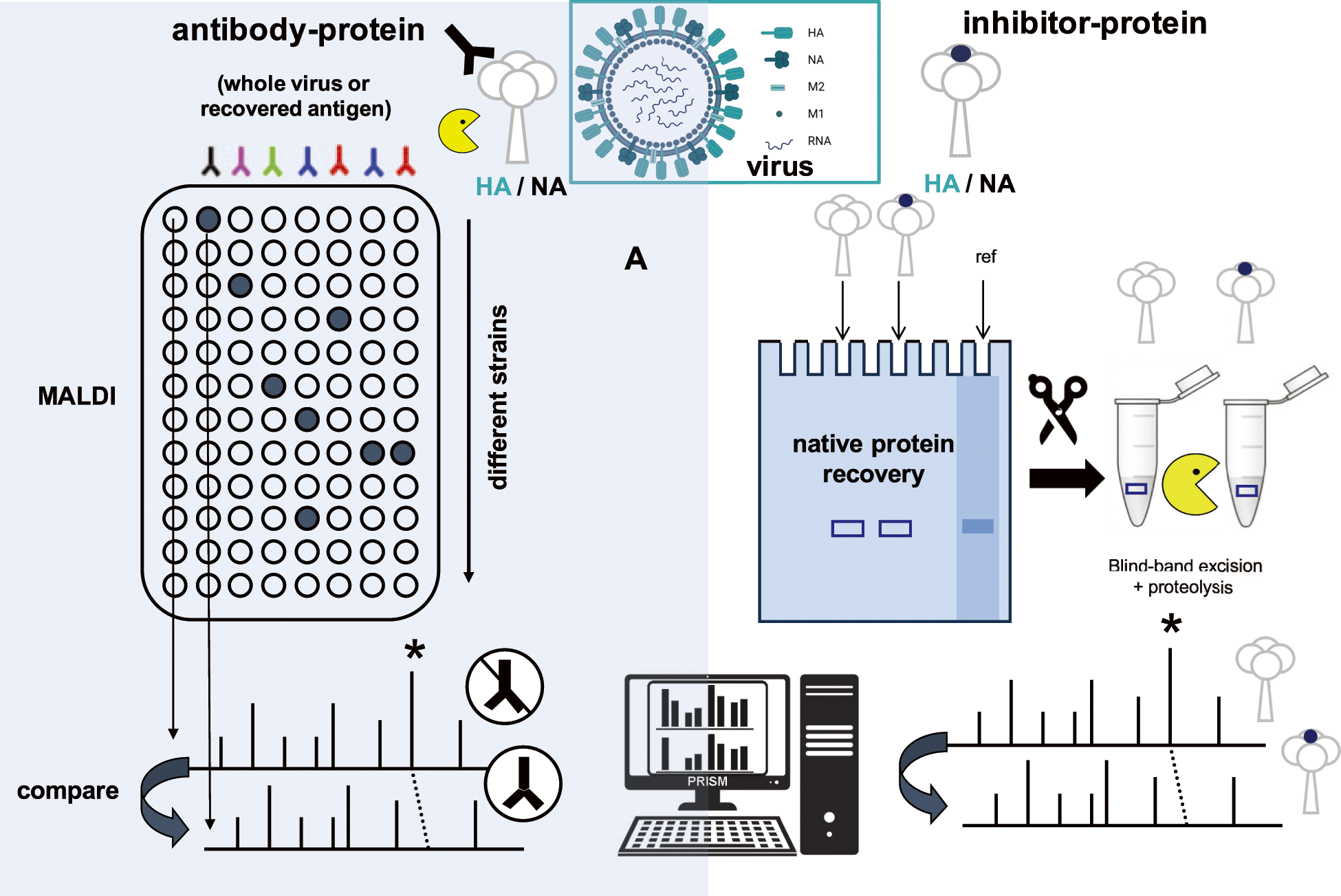
To characterise the antigenicity of a virus, a MALDI-MS-based immunoassay was developed17) that avoided the need to immobilise or recover the antibody–antigen complex ahead of analysis, which was a requirement of earlier approaches.36) While the MALDI mass map alone can be used to survey the virus at the protein level following proteolytic digestion of whole virus or its component antigens, the addition of a step in which one half of the mixture (post or prior to digestion) is treated with a monoclonal antibody and the remainder is left untreated.
A comparison of MALDI mass maps for antibody-treated samples over untreated (no antibody) controls demonstrated the selective reduction of peptide ions spanning the epitopic regions (Fig. 4A, left side). This also demonstrated, for the first time, that specific noncovalent antibody–peptide complexes could survive sample treatment, deposition, and the MALDI ionisation event.
A series of studies in which the epitopic sites, and thus the antigenicity, of viral hemagglutinin (HA) protein from common type A H1N1 and H3N2 subtypes of the influenza virus were subsequently reported.37–40) These studies utilised whole virus digests,37–40) as well as analyses of gel-recovered protein.41) Companion hemagglutination inhibition assays, in which the same antibodies were treated with red blood cells, were employed to validate the results.39)
The adoption of a time-course approach42) was also able to detect differences in the relative rates of binding of peptides, both from within and across epitopic domains, in noncompetitive and competitive experiments. A monoclonal antibody raised to target the HA1 subunit of the HA antigen of type A H3N2 influenza strains was found to recognize two epitopic peptides comprising HA residues 109–125 and 158–166 that likely form part of an extended discontinuous domain.42) Time-course experiments show that the smaller peptide binds antibody at a rate that is 5-fold faster than that for the larger peptide, while a subsequent study of modified synthetic variants of the former peptide was able to identify important antibody contact residues using this MS strategy.42)
To aid the above studies, an algorithm was written and compiled to run on a Windows operating system using a simple graphical user interface.43) The algorithm43) compares peak areas in a pair of MALDI mass spectra recorded for the control and antibody-treated sample to identify binding epitopic peptides based on measure of its reduction in absolute area measured as a percentage. In this regard, some tolerance (default 10%) is input by the user to take experimental fluctuations in mass spectrometric analysis into account. While developed for these specific purposes, the algorithm43) has general utility for the comparative analysis of differences within any two mass spectra.
The ability to preserve untethered or immobilised antigen–antibody complexes on MALDI surfaces led to the technique’s deployment to the study of antivirals (Fig. 4B, right side). A range of neuraminic acid and natural product inhibitors to influenza neuraminidase (NA) were studied to establish binding efficiencies and substituent effects.18,19,44) Native gel electrophoresis was used to separate a specific protein following virus denaturation, and one-half was treated with an inhibitor and ran alongside.18) After extraction from the gel, using a stained reference (ref) band, each was digested with protease. The two samples were then subjected side by side to MALDI-MS and the bound peptides identified based on their reduced peak area, as in the case of the antibody-treated protein/virus.
A natural product anthocyanidin inhibitor was found to bind within the so-called 430-cavity of the active site in the vicinity of NA residues 356–364 and 395–432,19) shielding proteases from releasing these peptide segments and thus reducing their detection. Importantly, this cavity is adjacent from the 150-loop region targeted by the inhibitor zanamivir within which resistance mutations have taken hold.
A subsequent study of related anthocyanidins,45) which differed in the number and position of the hydroxyl substituents on the phenyl group attached to the chromenylium ring, found subtle differences in their binding characteristics by MS that were in accord with their inhibitory properties assessed by neuraminidase inhibition assays. Companion molecular docking confirmed the results, while the differences in relative peak areas of the binding peptides across all compounds mirrored the half-maximal inhibitory concentration (IC50) measured in in vitro neuraminidase inhibition assays for each of compounds.
The binding of fusion inhibitors, Arbidol46) and an entry-blocker peptide,47) to influenza HA H1 and H5 subtypes was also investigated, together with parallel hemagglutination inhibition assays and molecular docking.46,47) The former inhibitor was found by MALDI-MS to bind to residues of 104–120 of the HA2 subunit, a region known to confer Arbidol resistance.46) Parallel molecular docking confirmed these results. The entry-blocker peptide was found by MS to bind in a reported sialic acid binding site surrounded by an α-helix (190-helix) and two-loop (130-loop and 220-loop) regions in the case of an H1 HA, and the second loop region in the case of an H5 HA.47) The peptide was shown to be able to maximally inhibit blood cell hemagglutination at a concentration of between 6.4 and 9.2 μM.
This strategy to detect and screen antiviral inhibitors, developed earlier for antigenicity studies, has broad applicability for any protein or other macromolecular complex that survives MALDI sample preparation, deposition, and the ionisation event. Indeed, others have applied the approach under an “intensity-fading MALDI” vernacular to a range of complexes.48) Our own studies suggest that noncovalent interactions with binding affinities down to the micromolar range (KD) are detectable.48)
Mass maps provide a unique signature for proteins that can be used to confirm their identity or establish differences with known proteins through searches of protein databases. Protein segments that maintain sequence conservation within viral types and subtypes allow for virus samples to be characterised accordingly. When high-resolution MS is employed, these peptides can be confidently and reliably identified by mass alone.
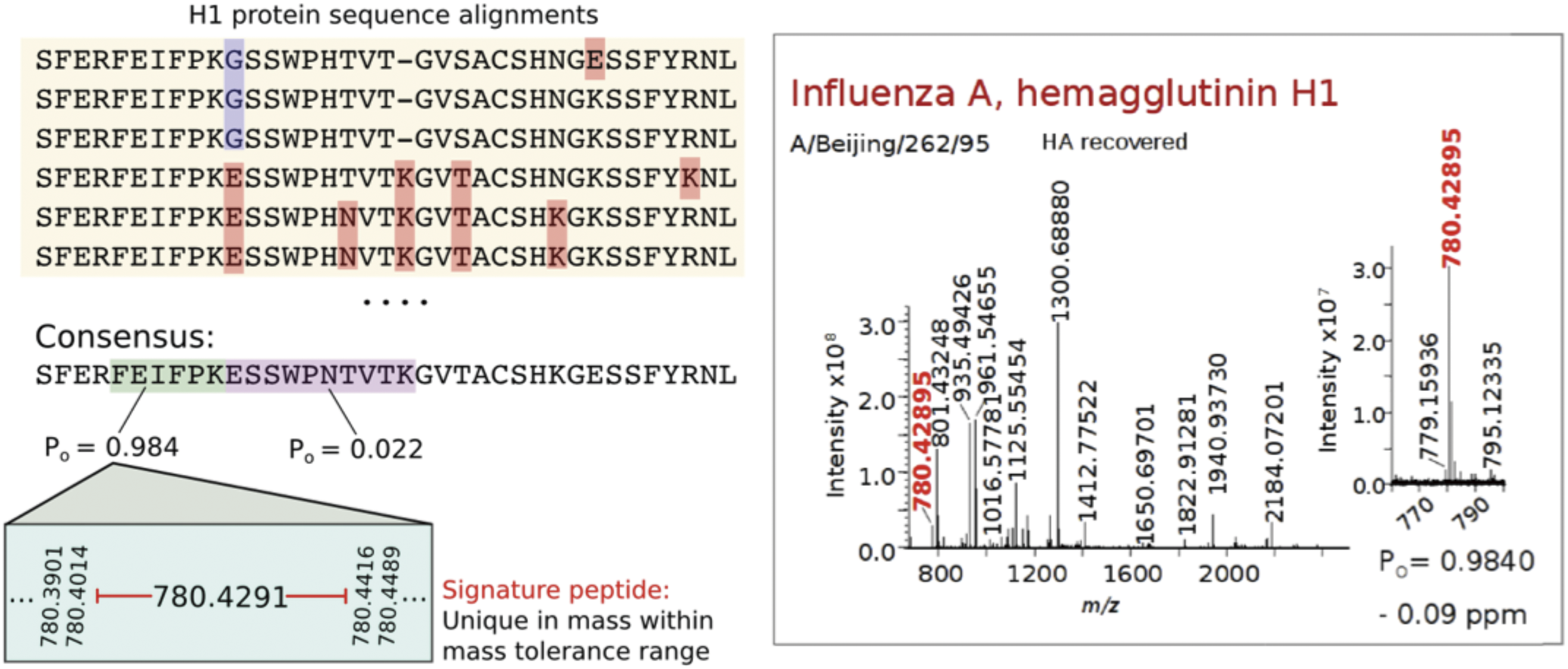
A proteotyping strategy20) was developed and applied to type and subtype influenza virus strains in whole virus and single viral protein digests through the detection of signature peptide biomarkers. These were first identified using specifically developed software,49) Translated gene sequences for each viral protein for a particular type, subtype and host were sourced from a sequence database. The sequences were aligned using the FluAlign algorithm49) and consensus sequences established to identify proteolytic peptides (produced from the digestion of a protein with a site-specific endoproteinase) that maintained high-sequence conservation. This was based on a measure of the frequency of occurrence (Po) of an amino acid at each residue within a peptide segment. The uniqueness of the theoretical monoisotopic masses of each signature peptide, against all proteolytic peptides across all known viral proteins, and possible contaminants such as albumins present in egg-grown viruses was then established using a second algorithm (FluGest), specifically designed for this purpose.49)
Signature peptides whose sequences were most conserved (Po≥0.95) and whose masses differed by greater than 5 ppm from all other proteolytic peptides enabled virus strains under analysis to be typed and subtyped with high confidence where one or more signatures were detected in a high-resolution mass spectrum. This is illustrated in Fig. 5 that shows an alignment of partial sequences for type A H1 HA in which mutations are highlighted. Of the two tryptic peptides in the consensus sequence below, one (FEIFPK) is highly conserved across all aligned sequences (with a Po=0.984) and is chosen as an H1 signature peptide. When detected in the spectrum of a whole virus or single protein digest (Fig. 5), the associated strain can be confidently identified to be of an H1 subtype (and thus type A H1N1 strain in humans).
The proteotyping strategy has been successfully implemented to type and subtype a wide range of strains across common human subtypes (type A H1N1 and H3N2, and type B influenza and parainfluenza) based upon the detection of signatures specific to each of the four viral proteins.50–57) Lineage-specific signatures were also identified and shown to be able to differentiate Victoria-87 from Yamagata-88 like strains.53)
Subsequent studies applied the same strategy to identify reassortment events given their prevalence in pandemic strains.58) A high-resolution MS study demonstrated the ability to distinguish pandemic from seasonal strains of the virus following viral reassortment that resulted in the outbreak of the 2009 H1N1 pandemic.59) Signature peptides of N1 neuraminidase were obtained for all type A H1N1, human H5N1, and 2009 pandemic H1N1 strains. Three signature peptides at m/z values of 549.2991, 1380.6287, and 2543.1296, unique to pandemic 2009 H1N1 strains, enable these strains to be differentiated from all other H1N1 and H5N1 strains in circulation at that time using high-resolution MS.
An alignment of translated HA gene sequences of all characterised type A H5N1 strains, or subsets thereof, enabled signature peptides for these strains to be determined from the perspective of the period upon which strains were isolated.60) Yet period-specific signature peptides were identified that enable strains associated with the 1997 H5N1 pandemic to be rapidly differentiated from those in circulation across the subsequent decade.
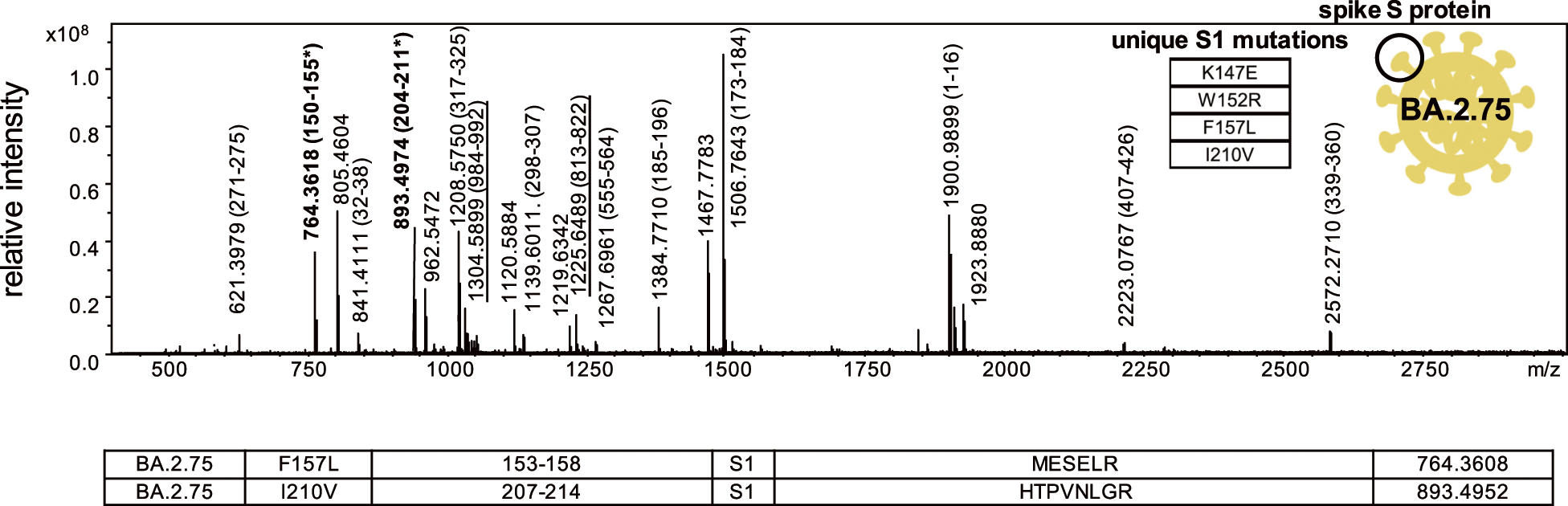
The proteotyping strategy has been applied to the SARS-CoV2 virus detected in clinical specimens.21,61–63) In these studies, the SARS-CoV2 virus across the five major variants of concern,62,63) as well as subvariants of omicron virus,21) has also been differentiated. To illustrate, two signature peptides associated with a common BA.2.75 subvariant with theoretical mass values of 764.3608 and 893.4952 (m/z [M+H]+ monoisotopic), associated with spike protein S1 subunit residues 152–157 and 207–214 containing mutations F157 and I210V, respectively, were detected in the spectrum of a recombinant form (labelled in bold in Fig. 6). These were subsequently in digested clinical specimens so as to enable strains of this subvariant to be identified.21)
The ability to measure peptide masses with accuracies of 5 ppm or better with a high-resolution mass spectrometer enables strains to be typed and subtyped in this manner from such mass-only measurements. Given multiple unique peptide mass signatures exist for the respiratory viruses studied, the detection of any number of them can be used to assign a variant with increasing confidence as more peptide masses match these values. Selected ion monitoring has been applied in a series of studies directed to the SARS-CoV2 virus to detect these signature peptides in clinical specimens to improve the detection limits and thus sensitivity of analysis.21,61–64)
No tandem mass spectrometric (MS/MS) sequencing is required, thus conferring both a sensitivity and time-saving benefit. While the sensitivity of low-resolution MALDI ToF instruments is superior by an order of magnitude or so with viruses identified at the 103 copies/mL level,65,66) the confidence in the peptide assignment is lower than for high-resolution mass spectrometers. This demonstrates the need for high mass resolution and accuracies to better discriminate SARS-CoV-2-positive and -negative samples.
The same high mass accuracy peptide mass maps can further be used to both chart and interrogate viral evolution.22–24) The masses of peptides produced after the proteolysis of a protein reflect the sequence of that protein. The more peptide masses that match from one protein to the next, the more homologous are the two proteins. Proteins containing the highest number of common masses within a specified mass error are grouped onto the same clade of a phylogenetic tree. The branch lengths reflect the ratio of number of common masses of the total in such sets.22) Neither the protein sequences themselves nor their alignment is required to build the tree, thus offering a truly “sequence-free” phylogenetic method.23)
Over the past decade, we have developed and applied this protein mass-based approach to both chart and study the evolution of viruses.22–24,58,67–74) Referred to as phylonumerics, it has also been shown that it can reliably identify and track single-point mutations68) in relation to that evolution to allow for the study of compensatory and resistance mutations69–71) by identifying single-point mutations from the difference in the masses of peptides derived from two homologous proteins. Such mutations are calculated during tree (“mass tree”) construction and displayed at various branch node positions.68) Beyond the study of viruses, the approach has been demonstrated to have universal versatility to resolve a wide range of organisms across the biological kingdoms of life75) and among animal species.76)
A mass tree produced from the peptide masses derived from the double digestion (trypsin+GluC) of the surface S-protein of the SARS-CoV2 virus for five major variants is shown in Fig. 7.24) The tree clearly resolves the variants based on peptide masses, which differ among each due to spike protein mutations within them. This tree and a much larger one (Fig. 8) built from mass data for some 3175 spike proteins display the same tree topology to those built from alignment of the protein sequence data through a visual comparison and the use of tree comparison algorithms.23)

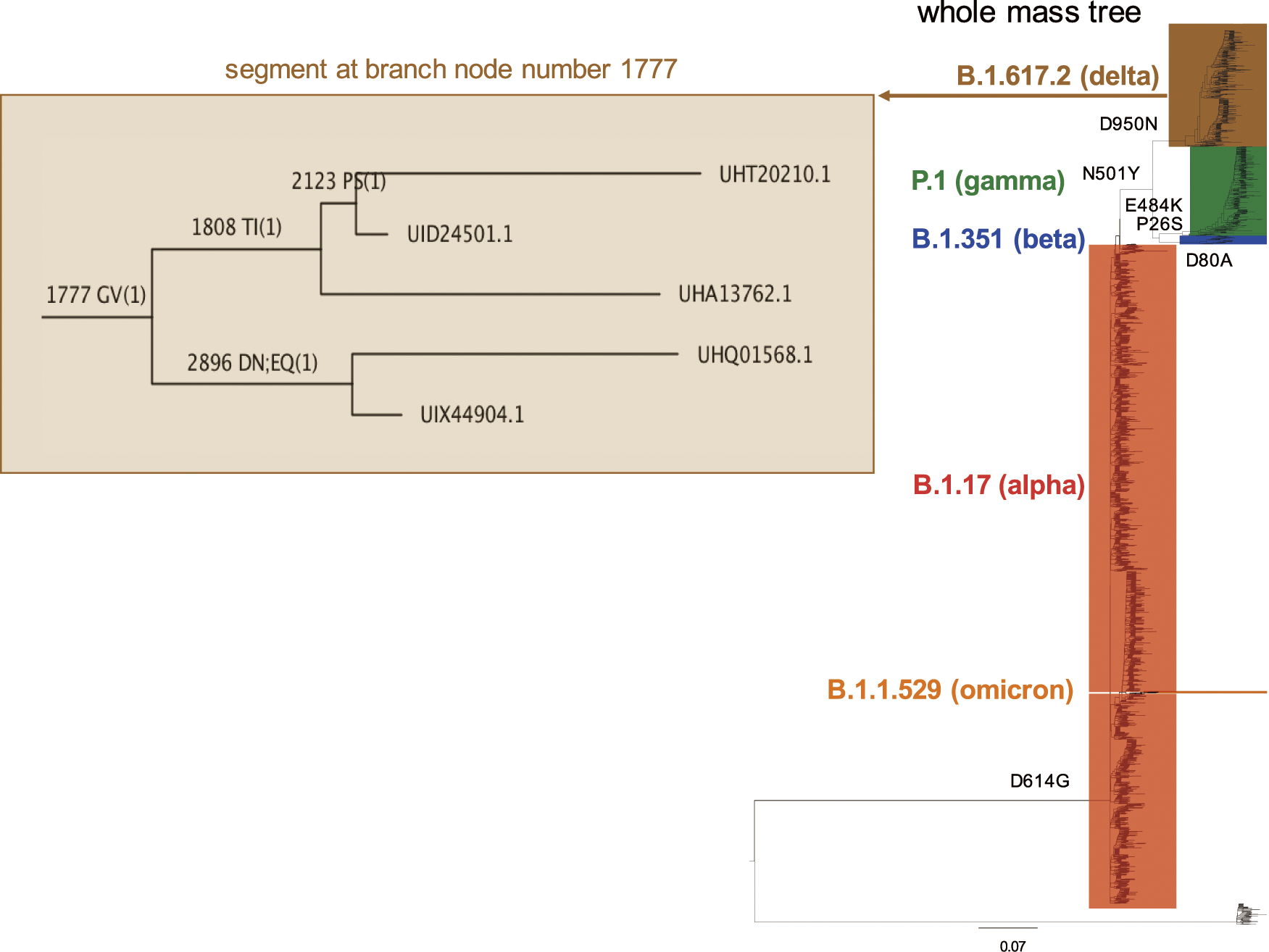
A segment of large mass tree spanning strains for the B.1.617.2 delta lineage (at branch node 1777) shows single-point mutations calculated by the mass tree algorithm that distinguishes the proteins labelled at the branch tips.23) The mass tree resolves the spike protein (accession UHT20210.1) of one strain from another (accession UID24501.1) based on the presence of a PS mutation. A comparison of the protein sequences of each reveals a single P to S mutation at position 249. These two proteins are diverged from protein UHA13762.1 at branch 1808 due to a TI mutation associated with a single TI mutation at position 1025. The three proteins are diverged based on the mass data alone from two other proteins at node 1777 due to a GV mutation. A comparison of the protein sequences with accessions UHA13762.1 and UHQ01568.1 reveals that they differ by the TI mutation and a GV mutation at position 1165.
Thus the mass trees22–24,58,67–76) not only correctly resolve and display different virus strains of different variants from one another, they also correctly identify (in some 80% of cases) mutations associated with viral evolution without the need for, or alignment of, the protein sequences themselves. This affords time saving and removes a computationally complex step associated with traditional gene or protein-based phylogenetic methods.77) Being protein focussed, it is more transferable to proteomics and structural biology methods, including those associated with drug design78) where an understanding of mutations that may hinder or benefit drug action is of interest.
The power of MS to both detect and identify, and distinguish major virus variants, as applied more recently to the SARS-CoV2 virus, is demonstrated. MALDI-MS, in particular, given its sensitivity, speed, and high sample throughput, should play a greater role in the frontline molecular detection of respiratory viruses such as influenza and the SARS-CoV2 virus in the near future, and represents an important and viable alternative to PCR-based detection methods.79,80) Although RT-PCR can achieve virus detection with few copies (~100 copies/mL), it requires at least 103 copies/mL for sequencing purposes. Total analysis times take at least 24–48 h due to the many biochemical steps involved and suffer from a relatively low-throughput capacity. Furthermore, due to the continual evolution of a virus, primers need to be continuously monitored for their effectiveness. If a probe or primer fails to bind, a PCR approach is rendered ineffective.
A high-resolution MALDI-MS approach offers benefits in terms of mass accuracy and confidence in virus identification and assignment, as reviewed in our own studies,81) and those using alternate instrument configurations.82) Indeed, the work conducted in Japan to build high-resolution compact mass spectrometers, employing extended flight tube and multi-pass time-of-flight configurations,83–85) has considerable merit and applicability to such studies.82) High-resolution instruments easily resolve peptides in whole virus digests, which feature multiple viral proteins and contaminants. Proteotyping viruses, using mass signatures alone, offers a considerable time saving over MS/MS strategies that consume more time and require at least an order of magnitude more sample. The use of high-resolution mass maps to build phylogenetic-like trees, and chart and study viral evolution affords an additional analytical benefit from such datasets.
The author thanks organisers of the 3rd International Biological Mass Spectrometry (iBMS) Symposium, Kyoto, October 2023 and members of the Mass Spectrometry Society of Japan (MSSJ) for the opportunity to present his work on which this paper is based. He particularly thanks Professor Susumu Uchiyama (Osaka University, Osaka, Japan) and Dr. Yuzo Yamazaki (Shimadzu Corporation, Kyoto, Japan) for facilitating this presentation. He acknowledges support from the Australian Research Council and the Clinical Research Fund for the work cited herein. He thanks current and former members of his laboratory who contributed to these studies, who are named co-authors on the many cited publications.
The author declares no competing financial or other conflict of interest in relation to this paper.
Based on an invited talk presented at the 3rd International Biological Mass Spectrometry (iBMS) Symposium of the Mass Spectrometry Society of Japan (MSSJ), October 2023.
Mass Spectrom (Tokyo) 2023; 12(1): A0136