INTRODUCTION
Good afternoon Ladies and Gentlemen,
Let me first thank the organizing committee of the 19th International Mass Spectrometry Conference (IMSC) for the invitation to present a tutorial lecture on gas-phase ion chemistry. This area has received much research interest since the mid fifties of the last century and has promoted many instrumental and methodological developments to obtain a detailed insight into the chemical behaviour and properties of gas-phase ions being responsible for the appearance of mass spectra. In the past two decades the focus of research has been shifted increasingly to life sciences, where medium-sized to very large molecules, such as in metabolomics, proteomics and genomics, are studied by use of not only newly developed instruments and methods, but also a number of techniques applied in gas-phase ion chemistry studies. The latter cover a very wide field, so that a selection of topics has to be made for presentation within the time allotted to this lecture. For this reason the author has chosen to discuss both old and more recent examples of ion chemistry studies involving:
1. The McLafferty rearrangement and tautomeric ion structures.
2. The determination of tautomeric ion structures, generated under electrospray ionization conditions, by application of gas-phase infrared multiphoton dissociation spectroscopy.
3. Time-resolved MS/MS.
4. The mechanism of epimerization of bis-Tröger bases, elucidated by ion mobility.
With regard to the methods used, only their essential characteristics will be mentioned where appropriate in order to focus as much as possible on ion chemistry. Then, in the electron ionization (EI) studies discussed it should be remembered that ionization takes place within 10−16 s, vibration times of the ions are 10−13–10−14 s, residence time of ions in the ion source is approximately 10−6 s and metastable ions, which can be studied in the field free regions of sector mass spectrometers during their flight on to the detector, have lifetimes of 10−5–10−6 s. Finally, because of the many crossing points in the potential energy surface, excited states, initially populated upon EI, are converted rapidly by radiationless transitions into vibrationally excited molecular ions in the electronic ground state, which plays a major role in their subsequent fragmentations.
The McLafferty rearrangement and tautomeric ion structuresThe intra-ionic tautomerization of a primary nitro group into its aci-nitro formOne of the most known reactions in mass spectrometry is the McLafferty rearrangement. First suggested for aliphatic aldehydes in 1956,1) the reaction involves a 1,5-γ-hydrogen atom shift to the radical/charge site at oxygen to form a protonated carbonyl group. This step is then followed by a homolytic cleavage of the C(α)–C(β) bond to give an enolic radical cation under expulsion of an olefin. The enolic radical cation is more stable than its keto-tautomer and they are separated from each other by a relatively high energy barrier for a suprafacial and orbital symmetry forbidden 1,3-hydrogen shift to effect tautomerization.2) After a long debate in the literature it is now well accepted that the McLafferty rearrangement proceeds in a stepwise fashion, while it can occur for a great variety of compounds, including ketones, acids, esters, amides, aromatic and so on. During his master’s degree study the author showed by site-specific D-labelling and use of a single focusing AEI MS2H mass spectrometer that the McLafferty rearrangement is also operative in the elimination of ethylene from the molecular ion of 1-nitropropane, yielding the radical cation of nitromethane in its tautomeric aci-nitro form with m/z 61. Although not stressed in the corresponding publication3) this reaction was depicted as a stepwise process because the molecular ion also eliminates a hydroxyl radical containing exclusively a hydrogen atom from the terminal methyl group. With use of the results obtained for 1-nitropropane and the notion that the molecular ion of methyl butyrate generates an abundant McLafferty rearrangement ion m/z 74 it was then decided to investigate the EI behaviour of the methyl ester of γ-nitrobutyric acid. Is it the nitro group or the ester group of this molecule which would direct the McLafferty rearrangement? Surprisingly, the peaks at both m/z 74 and m/z 61 appeared to be vanishingly small in its mass spectrum. Instead, elimination of HNO2 from the molecular ion was observed which is common for secondary and tertiary nitro compounds due to thermal decomposition, but not for primary nitro compounds which lose a NO2 radical. D-labelling has shown that the lost HNO2 molecule contains one of the hydrogens of its adjacent methylene group, suggesting an aci-nitro form of the corresponding molecular radical cation. However, this HNO2 loss cannot be due to ionization of the neutral compound in its aci-nitro form, because the molecular ions of the homologue compounds O2N(CH2)nCOOCH3 with n=1, 2, and 4 do not eliminate HNO2. They lose a NO2 radical in line with containing a primary nitro group. Following the HNO2 loss for n=3, a methyl radical is eliminated which fully originates from the ester group. On the basis of these observations Scheme 1 has been designed to account for the HNO2 loss from the molecular ion of the methyl ester of γ-nitrobutyric acid4):

Scheme 1.
First the radical/charge is located at the carbonyl oxygen as the ionization energy (IE) of a methyl ester is lower than that of a nitro group (IE of methyl butyrate=10.07 eV; IE of 1-nitropropane=10.81 eV5)). In the corresponding ion 1 a 1,5-γ-H shift can then take place to give ion 2, containing a protonated ester group and having a radical at the γ-carbon which is stabilised by the adjacent nitro group. This may then be followed by an estimated less than 71 kJ/mol endothermic proton transfer from the ester group to the nitro group (PA of methyl butyrate=836.4 kJ/mol; PA of nitroethane=765.7 kJ/mol5)) generating ion 3. In this way the nitro group is transformed into its tautomeric aci-form by virtue of the ester group, acting as a catalyst in the intra-ionic tautomerization. It is a route, which avoids the transformation of the nitro group into its aci-form by a direct suprafacial 1,3-hydrogen shift, being an orbital symmetry forbidden6) and consequently a high energy requiring reaction.
Attack of the radical on the carbonyl group in ion 3 would then lead to formation of the five-membered ring ion 4, from which HNO2 can be lost to give ion 5, where the charge can be delocalized over the adjacent oxygen atom by mesomerism and the radical site can induce elimination of the methyl radical by homolytic cleavage of the O–CH3 bond to yield ion 6. The latter reaction is unusual for methyl esters, but can be understood as a logical final step of the presented reaction sequence in Scheme 1.
The phenol and 1,3-cyclohexadien-5-one tautomersThe phenol radical cation can of course be generated by EI of phenol itself, but is it possible to generate its 1,3-cyclexadien-5-one tautomer via a McLafferty rearrangement in a suitable precursor ion? This question has been addressed by investigating the behaviour of the fragment (M–CH2CD2)·+ ion from the molecular ion of phenyl 1,1,1-trideuteroethyl ether. If a 1,5-D shift to the ortho position of the phenyl ring occurs to form the fragment ion, then the resulting mono-deuterated 1,3-cyclexadien-5-one ion is expected to transfer not only a deuteron, but also a proton to a base. The corresponding experiment, carried out in a drift cell ion cyclotron resonance mass spectrometer, has shown that the (M−CH2CD2)·+ ion exclusively transferred a deuteron, indicating that the phenol ion and not its 1,3-cyclexadien-5-one tautomer is formed.7) Since in 1980 very solid evidence has been presented for the occurrence of stable ion/molecule complexes during the unimolecular dissociation of ions,8) the formation of the phenol ion from phenyl 1,1,1-trideuteroethyl ether is best explained by the mechanism presented in Scheme 2. Upon stretching the O–CH2 bond in ion 7, the 1,1,1-trideuteroethyl cation in ion/molecule complex 8 will become spontaneously a deuteron π-bonded 1,1-dideuteroethylene ion, being a minimum energy structure9) to give ion 9, followed by deuteron transfer to oxygen to generate the phenol-OD ion 10 under expulsion of 1,1-dideutero-ethylene. This deuteron transfer reaction is exothermic by the amount of 192.9 kJ/mol (PA of C6H5O·=873.2 kJ/mol10); PA of C2H4=680.3 kJ/mol5)).

Scheme 2.
Following this result it was then decided to replace one of the methyl hydrogens in phenyl ethyl ether by a halogen atom expecting that this would favour a 1,5-hydrogen shift in the molecular ion to one of the ortho positions of the phenyl ring because of the possible repulsion between the electronegative oxygen and halogen atoms. 2-Phenoxyethyl chloride has been studied most extensively by a variety of methods and its molecular ion eliminates chloroethylene indeed to generate the C6H6O·+ ion. Specific D-labelling of each of its methylene groups has shown that the corresponding molecular ions lose in practically equal abundance CH2Cl and CD2Cl upon EI without any exchange with the aromatic ring hydrogens. For this phenomenon three intermediates have been considered11): a phenoxy radical/chloronium ion complex, a phenyl cationized ethylene oxide/chlorine atom complex and a conventionally bonded resonance hybrid structure where the phenyl ring and chlorine atom interchanged their position concertedly. The first was rejected because of the absence of radical stabilizing effects by appropriate substituents at the ring and the second was rejected because of the absence of the (M−Cl)+ peak in the EI spectrum, thus leaving the third one as the option. It should be noted that the positional interchange mentioned is not due to a thermal process, but really occurring in the molecular ion as shown by use of field ionization kinetics.12) This method enables to study in a time-resolved way unimolecular dissociations of molecular ions as a function of their lifetime from picoseconds to microseconds. At short molecular ion lifetimes of 2-phenoxyethyl chloride the methylene groups appeared to have retained their identity. Returning now to the formation of the C6H6O·+ ions, it might well be that part of them is generated from the earlier rejected phenoxy radical/chloronium ion complex in view of the fact that stable ion/molecule complexes during unimolecular dissociation can occur. However, the C6H5DO·+ ions from each of the D-labelled methylene 2-phenoxyethyl chlorides transfer for ∼65% a deuteron and for ∼35% a proton to a suitable base.13) The proton must originate from the phenyl ring, which is confirmed by the 43% deuteron transfer from the C6D5HO·+ ions of ring-d5 labelled 2-phenoxyethyl chloride. The question is whether this participation of the ring protons points to a 1,3-cyclexadien-5-one ion structure and thus to the aimed 1,5-hydrogen shift.
To this end the metastable decomposition behaviour of the C6H6O·+ from 2-phenoxyethyl chloride (and also from phenyl ethyl ether) has been compared to that of ionized phenol itself and its tautomeric 1,3-cyclexadien-5-one ion, being generated from ionized bicyclo[2,2,2]-oct-2-en-5,7-dione by ketene loss.
The molecular ion of phenol eliminates carbon monoxide and the reaction barrier height is 3.2 eV for decomposition in the ion source (at times ∼10−6 s), but 2.4 eV for decomposition in the metastable time frame of 10−5–10−6 s.
This means that the ions decomposing in the ion source are considerably more excited above the actual energy barrier due to a so-called kinetic shift than the metastable decomposing ions. This excitation or non-fixed internal energy may be released as kinetic energy of the fragments which is revealed and can be estimated from the broadening of the accompanying metastable peak. Upon lowering the ion accelerating voltage from 6 keV to 1 keV and thus increasing the phenol ion lifetime the mentioned kinetic energy release drops indeed and approaches the value observed for the CO loss from the metastable decomposing and tautomeric 1,3-cyclexadien-5-one ion.14) This behaviour being very characteristic for phenol ions has been observed also for the C6H6O·+ ions from phenyl ethyl ether and 2-phenoxyethyl chloride, indicating that most of those from the latter, if not all, have the phenol structure.15) However, this result does not exclude the aimed 1,5-H shift, because in that case the halogen atom may well abstract the proton from the ring and subsequently transfer it to the oxygen prior to elimination of chloroethylene, as visualized in Scheme 3.

Scheme 3.
Yet, collisional activated studies of the long-lived C6H6O·+ ions from 2-phenoxyethyl chloride have suggested that they have the tautomeric 1,3-cyclexadien-5-one ion structures16) like the earlier drift cell ion cyclotron resonance study.13) This has been confirmed eventually by a photodissociation study in the UV region, in which the corresponding spectra of authentic phenol and 1,3-cyclohexadien-5-one ions, being essentially different from each other, have been compared with the C6H6O·+ ions from phenyl ethyl ether and 2-phenoxyethyl chloride.17) Apparently, in ions 13 of Scheme 3 there is a competition between homolytic cleavage of the O–CH2 bond, generating the 1,3-cyclohexadien-5-one ion, and proton abstraction from the ring by the halogen atom to give eventually the phenol ion. In any case, it has been proven that in the molecular ion of 2-phenoxyethyl chloride a 1,5-H shift to one of the ortho positions of the phenyl ring occurs. Interestingly, it can also be concluded that conventional ion structures play a role in the elimination of chloroethylene from ionized 2-phenoxyethyl chloride notwithstanding the fact that in many mass spectrometric studies ion/molecule complexes are essential intermediates during unimolecular dissociations, which have made the borderline between unimolecular and bimolecular chemistry in the gas phase very thin indeed.
Anionic tautomers studied by gas-phase infrared multiphoton dissociation spectroscopyAn interesting example of negatively charged tautomeric ions concerns those of deprotonated p-hydroxybenzoic acid. Although in the gas phase benzoic acid is more acidic than phenol, p-hydroxybenzoic acid is even more acidic than either of them (ΔHoacid benzoic acid=1408.8 kJ/mol; ΔHoacid phenol=1451.4 kJ/mol and ΔHoacid p-hydroxybenzoic acid=1392.0 kJ/mol18)). The higher acidity of p-hydroxybenzoic acid is due to the larger delocalisation of the negative charge over the ring and carboxyl group when the phenolic proton is removed compared with removal of the carboxylic proton, where the negative charge is only delocalised over the carboxylic oxygens. A couple of years ago the group of Oomens of the FOM Institute for Plasma Physics Rijnhuizen in Nieuwegein, The Netherlands, which has moved recently to the Radboud University in Nijmegen, The Netherlands, posed itself the question which tautomer of deprotonated p-hydroxybenzoic acid would appear in the gas phase upon negative ion electrospray from protic and aprotic solvents.19) The carboxylate anion is favoured in protic solvents because of hydrogen bonding with the surrounding solvent molecules, whereas in aprotic solvents the phenolic anion tautomer would be preferred. Are these anion tautomer structures retained upon the transition from the liquid phase to the gas phase during the electrospray process? To answer this question the Free Electron Laser for Infrared eXperiments (FELIX), developed and built at the earlier mentioned FOM Institute, has been used enabling to perform infrared multiphoton dissociation (IRMPD) spectroscopy of gas phase ions over the wavelength range between 500 and 2000 cm−1. 20) Figure 1 shows the IRMPD spectra of deprotonated p-hydroxybenzoic acid, sprayed from the protic solvents methanol and water (panel A) and the aprotic solvents acetonitrile and dimethyl sulfoxide (panel C), while in panels B and D at the B3LYP/6-311++G(2d,2p) level of theory calculated infrared spectra are given.19) The spectrum in panel A is obviously different from that in panel C. In the former strong symmetric and antisymmetric carboxylate stretch modes at 1305 cm−1 and 1630 cm−1 are observed as indicated by the vertical red lines, confirming that in the protic solvents the carboxylic acid group has been deprotonated. This is further corroborated by the infrared spectrum calculated for this anion that has been given in panel B and that agrees very well with the experimentally obtained spectrum in panel A. In contrast, the spectrum in panel C shows bands typical for carboxylic acid C=O stretch at 1680 cm−1 and in-plane carboxylic acid COH bending mode at 1015 cm−1 as indicated by the vertical blue lines. Panel C therefore must be the infrared spectrum of p-hydroxybenzoic acid, which has been deprotonated at the phenol group, confirming that in aprotic solvents deprotonation of this group has taken place. The calculated infrared spectrum for this ion is given in panel D and agrees well with the experimentally obtained spectrum in panel C. It therefore can be firmly concluded that in this case the different tautomeric anions, generated by negative ion electrospray from protic and aprotic solvents, retain their structures upon transition from the liquid to the gas phase. These results also mean a caveat in the sense that solvents may influence chemically the outcome of the electrospray process.

Fig. 1. IRMPD spectra of deprotonated p-hydroxybenzoic acid generated by electrospray ionization from protic (methanol and water, panel A) and aprotic (acetonitrile and dimethyl sulfoxide, panel C) solvents compared to calculated infrared spectra of the carboxylate (COO−, panel B) and phenoxide (O−, panel D) tautomeric ions. Reprinted with permission from J.D. Steill and J. Oomens. J. Am. Chem. Soc. 131: 13570–13571, 2009. Copyright 2009 American Chemical Society.
An EI study of deuterated 3-penten-2-ol-1,1,1-d3 has shown that its molecular ion CH3CH=CHCH(OH)CD3·+ eliminates CH3· and CD3· in the ratio of 3 : 2 without any mixing of the H and D atoms.21) This is a peculiar observation because CH3 is bonded to an sp2-carbon atom and CD3 to an sp3-carbon atom, the former bond being stronger than the latter. The CH3· and CD3· loss has therefore been studied by use of field ionization kinetics (FIK), which permits to follow the reaction as a function of the molecular ion lifetime from picoseconds (10−12 s) to microseconds (10−6 s). It was found that at 10−10.8 s CD3· and CH3· are lost in the ratio of 3 : 2, which becomes 1 : 9 at 10−9.6 s and 2 : 3 at 10−5.2 s.22)
This behaviour has been interpreted on the basis of Scheme 4.

Scheme 4.
The IE’s being 9, 9.78, and 9.56 eV for pentene-1, pentanol-2, and 3-penten-2-ol5) it is reasonable to locate the radical/charge of the molecular ion 14 at the double bond which by homolytic cleavage can eliminate the methyl group at position 1 to give the protonated crotonaldehyde ion 15. In competition a 1,2-hydride shift from position C-2 to C-3 can take place to give ion 16 which by a successive 1,2-H shift can be transformed into the enolic ion 17. The latter can then eliminate by homolytic cleavage the methyl group at position 5, yielding the protonated methyl vinyl ketone ion 18. In going to longer molecular ion lifetimes up to microseconds a 1,4-H migration can take place in ion 16 to give the molecular ion 19 of pentanone 2, which by loss of the methyl group at position 1 results in the formation of the butyryl ion 20.
In line with the deuterium labelling results, two maxima have been observed in the normalized rate curve for methyl loss from unlabelled ionized 3-penten-2-ol, one at 10−10.5 s and another at 10−10.1 s. Collision-induced dissociation experiments on the [M−methyl]+ ions generated at 10−10.5 s and at 10−10.1 s yielded MS/MS spectra which indeed matched with those of authentic protonated crotonaldehyde and methyl vinyl ketone, respectively (see Fig. 2), being a unique example of time-resolved MS/MS.23)
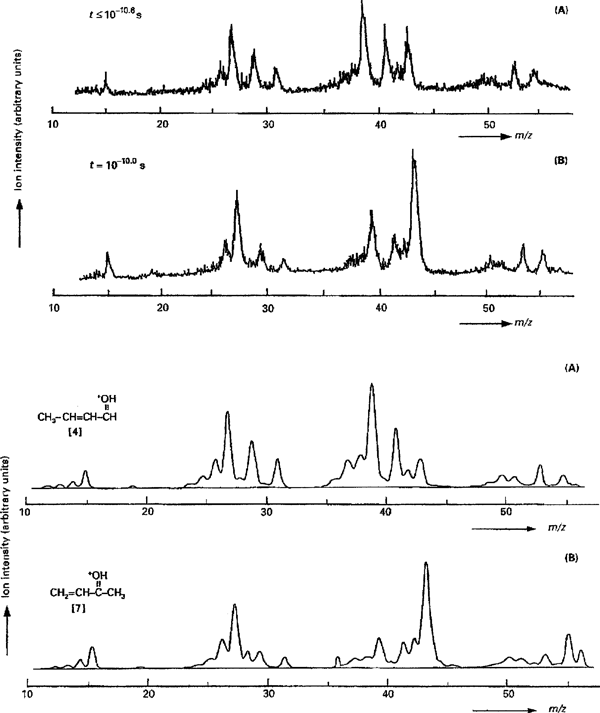
Fig. 2. The upper panels (A) and (B) show the collision-induced MS/MS spectra of the (M−methyl)+ ions, generated upon field ionization from the molecular ion of pent-3-en-2-ol at molecular ion lifetimes of ≤10−10.6 s and 10−10.0 s, respectively. The lower panels (A) and (B) show the reference collision-induced dissociation MS/MS spectra of protonated crotonaldehyde and protonated methyl vinyl ketone, respectively. Reprinted from Zappey et al. Org. Mass Spectrom. 26: 241–246, 1991 with permission from John Wiley and Sons Ltd., Copyright 1991.
Tröger’s base is a rigid concave-shaped molecule and contains two chiral nitrogen atoms, the 5S, 11S-enantiomer being shown below.
It was first synthesized by the German chemist Julius Tröger in 188724) but its structure was elucidated 48 years later.25) Tröger’s base and derivatives have found wide applications in different areas, such as supramolecular chemistry, molecular recognition, biological labelling, molecular tubes, catalysts, opto-electronics, receptors and ligands26–28) (and references cited therein).
Epimerization is a chemical process, where inversion of configuration at a single chiral center takes place in molecules containing more than one chiral center. Two mechanisms have been proposed for the pseudo-epimerization of Tröger bases29) which have been presented in Scheme 5 (note: because the chirality of both nitrogen atoms changes, pseudo-epimerization is in this case actually enantiomerization).

Scheme 5.
One of the mechanisms would proceed via a successive retro-Diels–Alder (RDA) and Diels-Alder (DA) reaction of the neutral 21→22→23 and vice versa. The other mechanism would proceed via N-protonation of 21 to give ion 24, which undergoes a proton catalyzed ring opening followed by ring closure via N–CH2 bond formation generating the ions 25 and 26, respectively. Deprotonation of the latter ion would then form the neutral 23 being the enantiomer of the neutral 21. To distinguish these two mechanisms Révész et al.30) have studied the bis-Tröger bases syn-1 and anti-1:
Epimerization of syn-1 leads to its diastereoisomer anti-1 and vice versa and these molecules have very different shapes, the former having a boat-like form and the latter a chair-like form. The same is true for N-protonated syn-1 and anti-1, the latter being calculated to be 2.1 kJ/mol more stable than the former. These differently shaped ions with identical m/z values could be separated from each other on the basis of their ion mobility31) in an inert buffer gas, the more compact N-protonated syn-1 boat-like ions having a higher ion mobility or lower cross section than the N-protonated anti-1 chair-like ions. For these experiments the SYNAPT G2 ion mobility mass spectrometer (Waters, Manchester, UK) was used, its order of components being ion source, quadrupole mass filter, linear ion trap, ion mobility device, transfer cell, time-of-flight mass spectrometer.32)
It has been shown that collisional activation of the N-protonated ions of pure syn-1 both in the ion source and in the ion trap in front of the ion mobility device leads to formation of the N-protonated ions of anti-1 and vice versa.
Similar experiments have been performed on the sodium cationized ions of syn-1 and anti-1, in which the sodium ion is coordinated to the π-electron system of the aromatic rings and for which calculations have shown that (syn-1+Na)+ is 39.7 kJ/mol more stable than (anti-1+Na)+. These ions could even be better separated from each other in the ion mobility device than their protonated analogues. However, neither pure (syn-1+Na)+ ions, nor pure (anti-1+Na)+ ions have shown any sign of epimerization following collisional activation. It therefore can be concluded that the epimerization of the bis-Tröger base studied proceeds via a proton-catalyzed ring-opening and not via a RDA reaction.30)

