Abstract
Owing to biotransformation, xenobiotics are often found in conjugated form in biological samples such as urine and plasma. Liquid chromatography coupled with accurate mass spectrometry with multistage collision-induced dissociation provides spectral information concerning these metabolites in complex materials. Unfortunately, compound databases typically do not contain a sufficient number of records for such conjugates. We report here on the development of a novel protocol, referred to as ChemProphet, to annotate compounds, including conjugates, using compound databases such as PubChem and ChemSpider. The annotation of conjugates involves three steps: 1. Recognition of the type and number of conjugates in the sample; 2. Compound search and annotation of the deconjugated form; and 3. In silico evaluation of the candidate conjugate. ChemProphet assigns a spectrum to each candidate by automatically exploring the substructures corresponding to the observed product ion spectrum. When finished, it annotates the candidates assigning a rank for each candidate based on the calculated score that ranks its relative likelihood. We assessed our protocol by annotating a benchmark dataset by including the product ion spectra for 102 compounds, annotating the commercially available standard for quercetin 3-glucuronide, and by conducting a model experiment using urine from mice that had been administered a green tea extract. The results show that by using the ChemProphet approach, it is possible to annotate not only the deconjugated molecules but also the conjugated molecules using an automatic interpretation method based on deconjugation that involves multistage collision-induced dissociation and in silico calculated conjugation.
INTRODUCTION
The conjugation reaction increases the hydrophilicity of a compound by attaching a hydrophilic molecule to it.1) An analysis of urine, one of the main destinations for excreted conjugates, reveals the presence of a wide range of conjugated compounds, originating not only from endogenous compounds but also from compounds derived from ingested food.2,3) In nontargeted metabolomics, a technique that is not limited to specific compounds, it is important to analyze these conjugates. Liquid chromatography coupled to mass spectrometry (LC/MS) is frequently used for the detection and structural analysis of conjugates because it has a higher sensitivity than NMR, despite the fact that it has a lower structural determination capability compared to NMR.4,5)
The product ion spectrum resulting from mass spectrometer data using collision-induced dissociation (CID) provides information regarding the substructure of a compound. However, manual data interpretation for spectrum annotation is a cumbersome task, requiring specific knowledge about mass spectrometry as well as the target compound. A computational approach can overcome these problems by virtue of its ability to provide an automatic interpretation.6–9) We previously reported on an automated annotation system for interpreting the nontargeted analysis of a multistage CID (MSn) spectrum, followed by the retrieval of candidates from a compound database.10) We were able to successfully annotate 20 components that showed contribution to tea quality, such as caffeine, catechins, and a series of organic acid esters. However, the identification of conjugates in nontargeted metabolomics remains a challenge. This is because the number of conjugated compounds in most databases is still limited. For example, a substructure search for compounds containing glucuronic acid in ChemSpider returned only 9,556 compounds, despite the fact that the database contains more than 32 million records (2014/9). Structural analyses of conjugates derived from known compound is used in studies of drug metabolism.4) Ridder et al. applied this approach to 75 known green tea components and generated 27,245 theoretical metabolites. As a result, they annotated 97 potential components, including 75 compounds that are not listed in PubChem.11) Because comprehensive information concerning both the ingested components and their metabolism is required, this technique cannot be easily applied to unknown components and metabolisms. Mass spectrometry following sample preparation through chemical or enzymatic deconjugation is also a known method for the structural analysis of and the identification of conjugates.12,13) Because the conjugated form cannot be directly identified using this method, a method that does not involve a deconjugation procedure is generally preferred.14)
In this study, we re-designed the compound annotation protocol and developed a method which we refer to as ChemProphet that can annotate compounds by taking into consideration conjugated forms using a compound database. ChemProphet uses an MSn spectrum to identify the type and number of conjugates, annotates the compounds in a deconjugated form based on a database search, and then evaluates the conjugated forms generated in silico.
We conducted compound annotation using a benchmark dataset in order to evaluate the performance of the ChemProphet protocol. We also analyzed chemical standards and confirmed the capability of our method to annotate the conjugates. Lastly, we annotated the conjugates present in the urine of mice that had been administered green tea. Green tea components15) and their metabolites have previously been identified using NMR16,17) and LC-MS12,16–20) analysis of authentic standards. We attempted to annotate these conjugated metabolites in a model experiment.
EXPERIMENTAL
Retrieval of candidates from the compound databaseFor a benchmark test, we retrieved candidates from PubChem by querying mono-isotopic mass, as shown in Table S1. We used ChemSpider for the other experiments. The candidates were retrieved by querying the predicted formulae. Duplicate structures and stereo isomers were eliminated.
Spectrum assignment, scoring, and rankingChemProphet searches for appropriate substructures whose molecular formula corresponds to the m/z value of the observed product ion. Rearrangements were not taken into consideration. ChemProphet calculates the final candidate score based on three different scores: the formula score, spectrum score, and penalty score. The penalty score is calculated based on the penalty value and the relative intensity Eq. (1).
 | (1) |
Where, single bond:
p=1, double bond:
p=2, triple bond:
p=3, bond including non-carbon:
h=1, carbon–carbon bond:
h=2, keton bond:
h=3.
The penalty value was calculated by Eq. (2).
 | (2) |
Where,
i ∈ Product ion.
PVi is minimum penalty value for each product ion.
RIi is relative intensity (%) of each product ion.
n is the number of product ion.
The rank of each candidate was determined by the final score. In the rank calculation, we used the “lowest rank,” determined as the sum of the number of candidates that showed a better score and the number of candidates that showed the same score. In this study, product ions with relative intensities of less than 5% were ignored.
In silico prediction of conjugatesConjugated forms are generated for selected candidates using the annotation results of a MS3 analysis. Specific groups involved in the conjugation were considered. Hydroxyl and amino groups were considered as substrates for sulfation, while hydroxyl, carboxyl, amino, and thiol groups were considered as substrates for glucuronidation.
Materials and analytical conditionsChemicals and reagents, sample preparation, instrumentation, and analytical conditions are described in Supplementary materials.
RESULTS AND DISCUSSION
Structure assignment and ranking using a benchmark datasetAlthough we previously used a commercial software program for predicting product ion, we developed ChemProphet equipped with newly designed automatic assignment and scoring procedure. The automatic assignment procedure was performed by predicting the formula of the observed product ion, a substructure search for matching to predicted formula, and an evaluation of neutral loss. As a result, the ratio of assigned product ion was improved, particularly in the case of a negative ion (data not shown). On the other hand, a holistic rise of assigned ion ratio resulted the need to an additional assessment in order to distinguish a preferable substructure. Therefore, we added a penalty score to evaluate the likelihood of bond dissociation. The penalty score was calculated based on the penalty value. The penalty value was proposed by Hill and Mortishire-Smith et al.,21) and a modified penalty value was also used by Ridder et al.9) A special penalty for a ketone bond was added for ChemProphet. We calculated the penalty score to normalize the difference in the maximum penalty value between the various candidates, and to consider the intensity of its product ion. The spectrum score was calculated in a manner similar to that for the structure score reported in a previous report.10)
Several computational annotation protocols have been reported.6–9) Hill et al. developed a protocol using Mass Frontier Ver. 4, and the evaluation involved the annotation of product ion spectra derived from 102 compounds.6) Wolf et al. developed the MetFrag protocol based on an annotation by comparing with computational fragmentation and scoring with the bond dissociation energy of the cleaved bond.8) Ridder et al. developed an annotation protocol referred to as MAGMa for MSn analysis using computational assignment and scoring based on a penalty value.9)
Our approach involved the use of the benchmark dataset reported by Hill et al. as well as Wolf et al. and Ridder et al. In order to compare the results, we generated a merged product ion spectrum and retrieved candidates using PubChem in a manner similar to a previous report.8) The results of the annotation are shown in Tables 1, S2–S5. Although we could not compare each result precisely, because the number of retrieved candidates was different, our protocol showed better results than previous reports in terms of average, median, and third quartile rank. The processing time of ChemProphet was more varied than the others (Figs. S1 and S2). The difference in time was caused by differences associated with the algorithm used for the assignment, because ChemProphet searched substructures from scratch for each formula.
Table 1. Statistical results of ranking the benchmark dataset in comparison to published studies.
| ChemProphet | Hill et al.a) | Wolf et al.a) | Ridder et al.b) |
|---|
| Product ion | Merged | Selected | Merged | Merged |
|---|
| Average rank | 14.7 (+/−3.6) | 44.2 (+/−14.1) | 24 (+/−7.9) | 30.8 (+/−9.8) |
| Std. deviation | 36.6 | 142.5 | 80.2 | 98.9 |
| Median rank | 3 | 4 | 4.5 | 3 |
| 3rd quartile rank | 8 | 17.5 | 11.75 | 9 |
a) Rank for each candidate were supplied as additional file published by Wolf et al.b) Rank for each candidate were supplied as supporting information published by Ridder et al.
The annotation process for the conjugates involves three main phases: 1. Prediction of the deconjugated formula based on an assessment of the type and number of possible conjugate reactions; 2. Assignment and ranking of the deconjugated candidates retrieved from the compound database; and 3. Evaluation of the in silico generated conjugates based on the ranking of the deconjugated form, as shown in Fig. S3. The type of conjugation can be recognized by a neutral loss.5) We automatically acquired the product ion spectra for the deconjugated ion using a data-dependent MS3 analysis triggered by the neutral loss observed in a MS2 analysis. The number of conjugate reactions can be determined by the depth of the data-dependent MSn analysis.
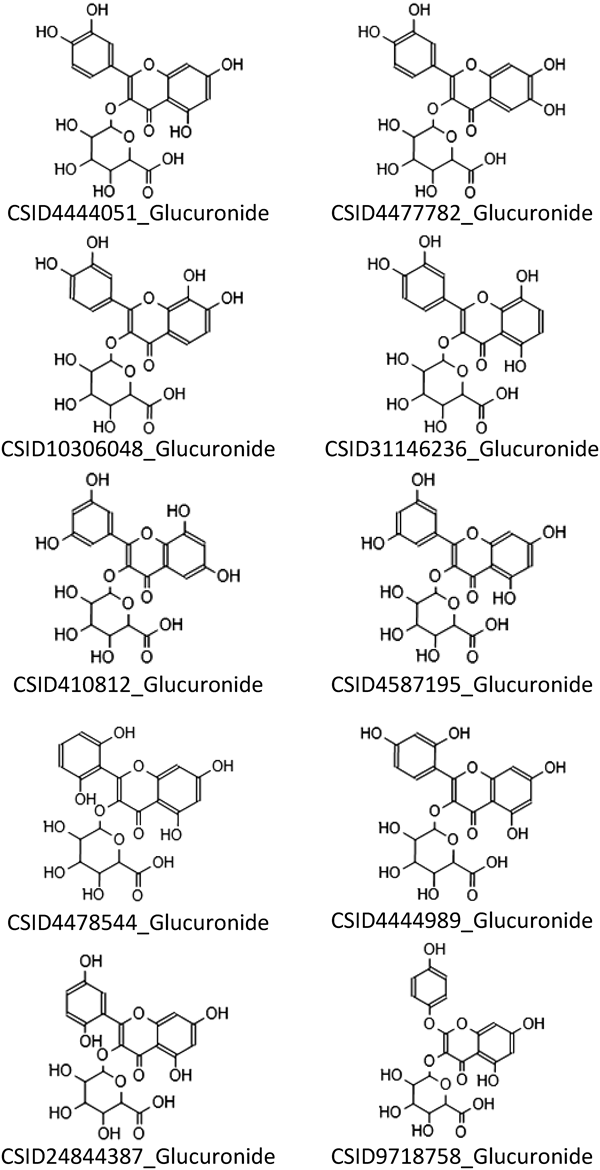
We analyzed the quercetin 3-glucuronide as a standard compound and annotated its MSn spectra to evaluate the capability of our protocol to annotate conjugates. In the MS2 spectrum, the deprotonated quercetin molecule was observed as a product ion at m/z 301.0354. Subsequently, the MS3 spectrum of quercetin was acquired automatically. A list of predicted formulae including the correct formula, C21H18O13, was obtained. ChemSpider returned 59 candidates by querying the deconjugated formulae. After annotation of the MS3 analysis with the retrieved candidates, quercetin ranked 15th. After annotation of the MS2 analysis with the in silico generated glucuronide conjugates, quercetin 3-glucuronide ranked 10th and nine candidates except for CSID 9718758 showed the same score (Fig. 1). These candidates had a similar backbone structure and the position of conjugation was the same.
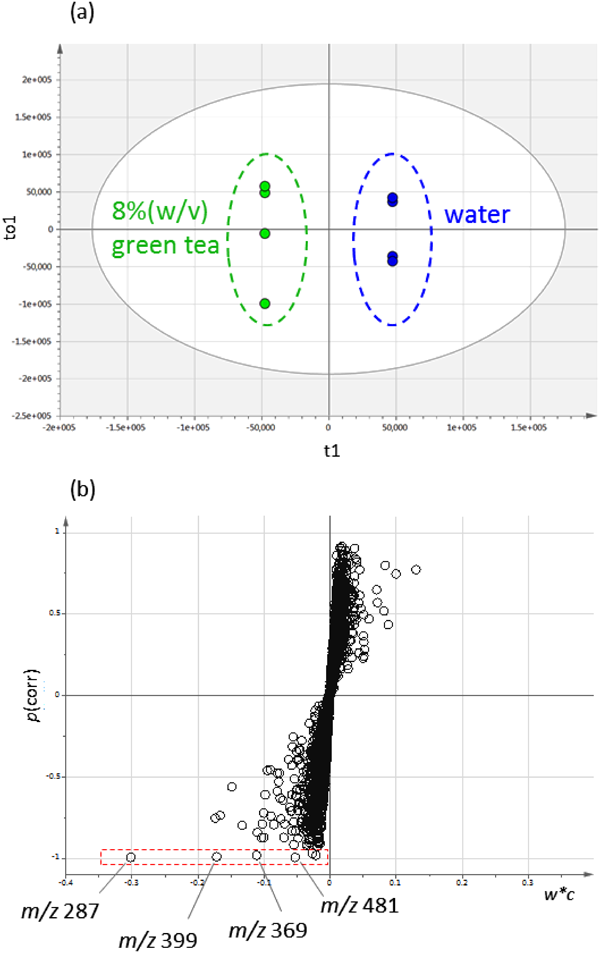
Analysis of conjugates of green tea components in mouse urineIn a model experiment, we analyzed the urine of mice that had been administered a green tea extract, and searched for urinary conjugates derived from the green tea components. We used the negative mode ESI because these conjugates have acidic moieties attached. Representative chromatograms are shown in Fig. S4. A total of 3,105 peaks were detected. An OPLS regression analysis was used to extract the components of the green tea and its metabolites (Fig. 2). An assessment of significant neutral loss and an S-plot showed that four peaks, m/z 287, 369, 399, and 481 corresponding to conjugates of green tea components.
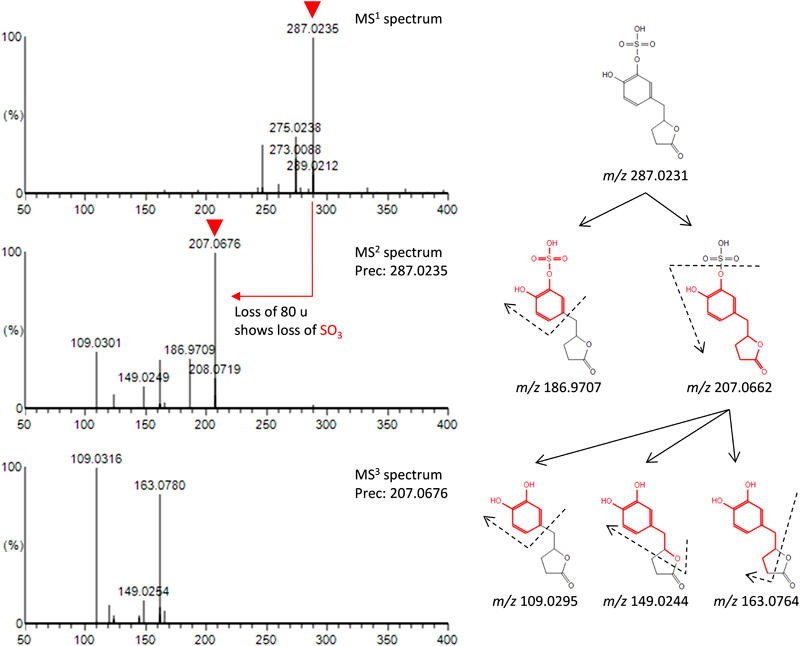
Table 2 provides a summary of the annotated candidates for these four features. Figure 3 shows the result of the assignment for di-hydroxyphenyl-γ-valerolactone sulfate. Here, the structure of the 3′-O-sulfate is shown, but the two positions assigned for 3′-O-sulfate and 4′-O-sulfate were not differentiated. Di-hydroxyphenyl-γ-valerolactone is a known metabolite that is produced by the C-ring cleavage of catechin. Li et al. identified di-hydroxyphenyl-γ-valerolactone-O-sulfate as a metabolite of green tea in human urine using NMR16) and mass spectrometry.18) Catechin-O-sulfate, O-methyl-gallocatechin-O-sulfate, and gallocatechin-O-glucuronide were previously reported as urinary metabolites derived from catechin in green tea by LC-MS analysis by comparison with an authentic standard and by NMR.12,16,18) As a result of the annotation of the generated sulfate conjugate, the scores for 3′- and 4′-O-sulfate were relatively high (Figs. S5 and S6). Romanov-Michailidis et al. synthesized the conjugated sulfate and detected it in human biological fluids.22) The product ion spectra of the synthetic sulfate conjugate showed that m/z 231 corresponds to the 3′- or 4′-O-sulfate, and the product ion at m/z 247 corresponds to the 5- or 7-O-sulfate.23) Compared to previous results, the assignment results for O-methyl-gallocatechin also suggest that a sulfate conjugated to the B-ring is produced. On the other hand, catechin-O-sulfate and gallocatechin-O-glucuronide were not reported by Ridder et al.11) The result of retrieving the annotated conjugate from ChemSpider is also shown in Table 2. No candidates were found in database.
CONCLUSION
Although compound databases are being improved each day, they are still incomplete in terms of information regarding conjugates. Thus, we developed a new system referred to as ChemProphet for annotating conjugates that are not found in databases. It also includes a newly designed automatic assignment and scoring process. We assessed the developed annotation protocol using a benchmark dataset and compared our results to those from previous studies. It was also demonstrated that ChemProphet can be used to annotate compounds by the annotation of quercetin 3-glucuronide and urinary conjugates derived from green tea components. This protocol can be applied to annotating conjugates derived from components of not only food and beverages2) but also other complex materials such as waste water24) in biological samples. In this report, we focused on glucuronide and sulfate groups as important urinary conjugates. However, there are other reactions worth investigating, such as glutathione conjugation, glycine conjugation, methylation, and acetylation. The ChemProphet approach can be applied to these conjugates in the same way, because it is known that these conjugate reactions also generate a specific neutral loss.5)
Acknowledgment
The study represents a portion of the dissertation submitted by Tairo Ogura to Osaka University in partial fulfillment of the requirement for his Ph.D.
REFERENCES
- 1) Casarett & Doull’s Toxicology. The Basic Science of Poisons, 7th ed. (Ed.: K. Curtis), McGraw-Hill, New York.
- 2) S. Bouatra, F. Aziat, R. Mandal, A. C. Guo, M. R. Wilson, C. Knox, T. C. Bjorndahl, R. Krishnamurthy, F. Saleem, P. Liu, Z. T. Dame, J. Poelzer, J. Huynh, F. S. Yallou, N. Psychogios, E. Dong, R. Bogumil, C. Roehring, D. S. Wishart. The human urine metabolome. PLoS ONE 8: e73076, 2013.
- 3) R. Llorach, M. Urpi-Sarda, O. Jauregui, M. Monagas, C. Andres-Lacueva. An LC-MS-based metabolomics approach for exploring urinary metabolome modifications after cocoa consumption. J. Proteome Res. 8: 5060–5068, 2009.
- 4) W. A. Korfmacher ed. Using Mass Spectrometry for Drug Metabolism Studies. 2nd ed. CRC Press, Florida, 2010.
- 5) K. Levsen, H.-M. Schiebel, B. Behnke, R. Dötzer, W. Dreher, M. Elend, H. Thiele. Structure elucidation of phase II metabolites by tandem mass spectrometry: An overview. J. Chromatogr. A 1067: 55–72, 2005.
- 6) D. W. Hill, T. M. Kertesz, D. Fontaine, R. Friedman, D. F. Grant. Mass spectral metabonomics beyond elemental formula: Chemical database querying by matching experimental with computational fragmentation spectra. Anal. Chem. 80: 5574–5582, 2008.
- 7) M. Heinonen, A. Rantanen, T. Mielikäinen, J. Kokkonen, J. Kiuru, R. A. Ketola, J. Rousu. FiD: A software for ab initio structural identification of product ions from tandem mass spectrometric data. Rapid Commun. Mass Spectrom. 22: 3043–3052, 2008.
- 8) S. Wolf, S. Schmidt, M. Müller-Hannemann, S. Neumann. In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC Bioinformatics 11: 148, 2010.
- 9) L. Ridder, J. J. J. van der Hooft, S. Verhoeven, R. C. H. de Vos, R. van Schaik, J. Vervoort. Substructure-based annotation of high-resolution multistage MSn spectral trees. Rapid Commun. Mass Spectrom. 26: 2461–2471, 2012.
- 10) T. Ogura, T. Bamba, E. Fukusaki. Development of a practical metabolite identification technique for non-targeted metabolomics. J. Chromatogr. A 1301: 73–79, 2013.
- 11) L. Ridder, J. J. J. van der Hooft, S. Verhoeven, R. C. H. de Vos, J. Vervoort, R. J. Bino. In silico prediction and automatic LC-MSn annotation of green tea metabolites in urine. Anal. Chem. 86: 4767–4774, 2014.
- 12) C. Li, X. Meng, B. Winnik, M. J. Lee, H. Lu, S. Sheng, B. Buckley, C. S. Yang. Analysis of urinary metabolites of tea catechins by liquid chromatography/electrospray ionization mass spectrometry. Chem. Res. Toxicol. 14: 702–707, 2001.
- 13) W. M. A. Niessen ed. Current Practice of Gas Chromatography-Mass Spectrometry (Chromatographic Science Series). Marcel Dekker, Inc., 2001, p. 519.
- 14) R. L. Gomes, W. Meredith, C. E. Snape, M. A. Sephton. Conjugated steroids: Analytical approaches and applications. Anal. Bioanal. Chem. 393: 453–458, 2009.
- 15) H. N. Graham. Green tea composition, consumption, and polyphenol chemistry. Prev. Med. (Baltim) 21: 334–350, 1992.
- 16) C. Li, M. J. Lee, S. Sheng, X. Meng, S. Prabhu, B. Winnik, B. Huang, J. Y. Chung, S. Yan, C. T. Ho, C. S. Yang. Structural identification of two metabolites of catechins and their kinetics in human urine and blood after tea ingestion. Chem. Res. Toxicol. 13: 177–184, 2000.
- 17) M. Harada, Y. Kan, H. Naoki, Y. Fukui, N. Kageyama, M. Nakai, W. Miki, Y. Kiso. Identification of the major antioxidative metabolites in biological fluids of the rat with ingested (+)-catechin and (−)-epicatechin. Biosci. Biotechnol. Biochem. 63: 973–977, 1999.
- 18) X. Meng, M. J. Lee, C. Li, S. Sheng, N. Zhu, S. Sang, C. T. Ho, C. S. Yang. Formation and identification of 4′-O-methyl-(−)-epigallocatechin in humans. Drug Metab. Dispos. 29: 789–793, 2001.
- 19) R. C. Pimpão, T. Dew, M. E. Figueira, G. J. McDougall, D. Stewart, R. B. Ferreira, C. N. Santos, G. Williamson. Urinary metabolite profiling identifies novel colonic metabolites and conjugates of phenolics in healthy volunteers. Mol. Nutr. Food Res. 58: 1414–1425, 2014.
- 20) H. Lu, X. Meng, C. Li, S. Sang, C. Patten, S. Sheng, J. Hong, N. Bai, B. Winnik, C. Ho, C. S. Yang. Glucuronides of tea catechins: Enzymology of biosynthesis and biological activities. Drug Metab. Dispos. 31: 452–461, 2003.
- 21) A. W. Hill, R. J. Mortishire-Smith. Automated assignment of high-resolution collisionally activated dissociation mass spectra using a systematic bond disconnection approach. Rapid Commun. Mass Spectrom. 19: 3111–3118, 2005.
- 22) F. Romanov-Michailidis, F. Viton, R. Fumeaux, A. Lévèques, L. Actis-Goretta, M. Rein, G. Williamson, D. Barron. Epicatechin B-ring conjugates: First enantioselective synthesis and evidence for their occurrence in human biological fluids. Org. Lett. 14: 3902–3905, 2012.
- 23) M. Dueñas, S. González-Manzano, F. Surco-Laos, A. González-Paramas, C. Santos-Buelga. Characterization of sulfated quercetin and epicatechin metabolites. J. Agric. Food Chem. 60: 3592–3598, 2012.
- 24) R. Al-Salhi, A. Abdul-Sada, A. Lange, C. R. Tyler, E. M. Hill. The xenometabolome and novel contaminant markers in fish exposed to a wastewater treatment works effluent. Environ. Sci. Technol. 46: 9080–9088, 2012.