Special Issue on Materials Science on High-Entropy Alloys
Doping of Interstitials (H, He, C, N) in CrCoFeNi High Entropy Alloy: A DFT Study
2020 Volume 61 Issue 4 Pages 616-621
Details
2020 Volume 61 Issue 4 Pages 616-621
The dissolution behavior of H, He, C and N in CrCoFeNi high entropy alloy (HEA) is studied by density functional theory calculation. Results show that the site preference of H, C and N in CrCoFeNi HEA is the octahedral site, while the site preference of He is the tetrahedral site. The dissolution energy of H in CrCoFeNi HEA is the lowest and that of He is the highest. The high dissolution energy of He is considered to be due to its closed-shell electronic structure. Moreover, results suggest that the H, He and N are more stable at the site with more Cr atoms, and C is more stable at the site with less Ni atoms. Furthermore, It is found that H, He, C and N have the effect in decreasing the magnetic moments of solutes in CrCoFeNi HEA. Electronic structure analysis shows that, for C and N, there is strong hybridization between them and solutes. It implies the chemical bonding between C, N and solutes is strong.

The doping of H, He, C and N in CrCoFeNi High entropy alloy.
High entropy alloys (HEAs), the alloys are based upon multiple atomic species in near equimolar ratios rather than a single principal element, has drawn much attention in recent years.1–8) Past works show that HEAs have some remarkable properties such as excellent mechanical properties,2,9–11) good corrosion resistance12,13) and high irradiation tolerance.5,14–16) Among all the reported HEAs, CrCoFeNi HEA system is the one which has been thoroughly studied by both experimental9,16–22) and theoretical22–26) methods.
Interstitial atoms like hydrogen (H), Helium (He), carbon (C) and nitrogen (N) are well known to have a significant impact on the mechanical properties of metals. Very recently, several research works focused on the doping effect of these interstitials on CrCoFeNi HEA family. It is found that H with appropriate amount can be utilized to tune beneficial strengthening and toughening mechanisms rather than undergoing catastrophic failure due to hydrogen embrittlement.27) C doping can increase the yield strength, ultimate tensile strengths as well as the plasticity of HEA.11) Moreover, N increases the hardness of HEA.28) These researches have proved that the doping effect of interstitial in CrCoFeNi HEAs is significant and particular. However, the solution behavior of these interstitial and how they affect the physical properties of CrCoFeNi HEA have not been completely understood.
In this work, the fundamental phenomena of H, He, C and N in CrCoFeNi HEA, and their effect on the physical properties, electronic structures of CrCoFeNi HEA are investigated with density functional theory (DFT) calculation.
All calculations in this work are performed with the program packages GPAW.29) The Generalized Gradient Approximation (GGA) PBE30) functional is used for exchange-correlation. The interaction between the valence and core electrons is modelled with projector augmented wavefunctions.31) A plane wave cut-off of 450 eV is applied together with a smearing of electronic states of 0.1 eV. In this work, CrCoFeNi HEA is considered to be solid solution with fcc structure. It is modelled with a (3 × 3) fcc supercell which containing 108 atoms, as shown in Fig. 1. A 3 × 3 × 3 k-point grid is utilized for sampling Brillouin zones. The randomness of Cr, Co, Fe and Ni in the lattice sites are achieved by implementing the special quasi-random structure (SQS) method, as used in previous work.22) Spin-polarization effects are taken into consideration for all calculations. Each structure is fully relaxed until the atomic force is less than 0.03 eV. Furthermore, for the calculation includes H atom, due to the small mass of H atom, zero-point vibrational (ZPV) energy is considered.
The schematic graph of CrCoFeNi HEA.
To achieve a large number of possible arrangements of HEAs, 6 random solid solutions of CrCoFeNi are created with SQS method. At first, the lattice constant of each CrCoFeNi HEA is checked. Results are shown in Table 1. The lattice constant of CrCoFeNi HEA is calculated to be 3.51 Å in average, which is in good agreement with former report.22,23) It is smaller than the experimental value,22) but the difference is only about 1%.

The magnetic property of CrCoFeNi HEA is calculated to be ferromagnetic, which is the same as previous reports.23,32) The magnetic moment of each solute is shown in Fig. 2(a). It can be seen that Fe, Co and Ni produce magnetic moment in the same direction with 1.87 µ, 0.95 µ and 0.26 µ in average, respectively. While the average magnetic moment of Cr is −0.66 µ. Most Cr atoms in CrCoFeNi HEA show the opposite magnetic moment against Fe, Co and Ni, but still, some Cr atoms produce the magnetic moment in the same direction as Fe, Co and Ni. Moreover, it is notable that all these four solutes show a variety in magnetic moment value. The variation in the magnetic moment of Cr is large, while the variation in the magnetic moment of Ni is small.

The magnetic moment (a) and charge transfer (b) of each solute in CrCoFeNi HEA.
Furthermore, the charge transfers of each solute in CrCoFeNi HEA are analyzed, as presented in Fig. 2(b). Clearly, in all the cases, Ni atoms have electrons positive charges and Cr atoms have negative charges. For Co atoms, in most cases they have positive charges, but a few Co atoms have negative charges. For Fe atoms, half of them have positive charges and another half have negative charges. This means that within CrCoFeNi HEA, Cr atoms play as the main electron donor, Ni atoms play as the main electron acceptor. For Co and Fe atoms, they can be electron donor or electron acceptor, which depends on the local configuration surround them.
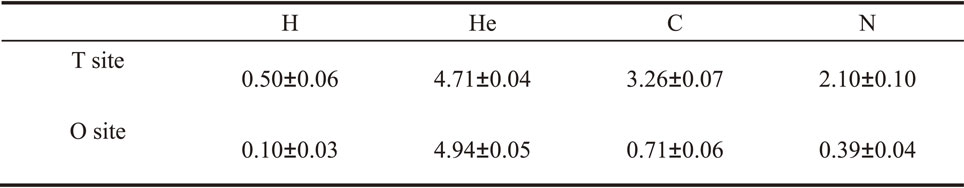
3.2 Dissolution of H, He, C and N in CrCoFeNi HEAThe dissolution behavior of H, He, C and N in CrCoFeNi HEA is studied. As first, the site preference of H, He, C and N is investigated by calculating the dissolution energy (Edis) of these interstitials at tetrahedral site (T site) and octahedral site (O site) in CrCoFeNi HEA, respectively. Considering that Edis may be different from site to site in CrCoFeNi HEA, 3 tetrahedral sites and 3 octahedral sites for each interstitial are considered to obtain the Edis.
The Edis is calculated by the following formula:
| \begin{equation} \mathrm{E}_{\textit{dis}} = \mathrm{E}_{\textit{tol}} - \mathrm{E}_{\textit{HEA}} - \mathrm{E}_{\textit{Inter}} \end{equation} | (1) |
Calculated average Edis of H, He, C and N in CrCoFeNi HEA are presented in Table 2. Results show that H, C and N atom at O site have lower Edis compared with they are at T site, indicating they are more stable at O site rather than T site. However, He is more stable at T site with lower Edis compared with O site. The result is similar to the case of H, He, C and N in Ni and fcc Fe as reported before.33,34)
In further study, to systematically investigate the dissolution behavior of H, He, C and N, they are put at the different sites in CrCoFeNi HEA, and the Edis is calculated. For each interstitial, 24 different interstitial sites are considered. According to the previous results, in these calculations, the occupation site of H, C and N is considered to be O site, while the occupation site preference of He is considered to be T site. Figure 3 shows the Edis of H, He, C and N at 24 different interstitial sites. It can be seen that H has the lowest average Edis, while He has the highest average Edis. Although the atom radius of C and N is much larger than it of He, the Edis of C and N is below it of He. It is noticeable that N has a lower Edis than C in CrCoFeNi HEA. Former researches also show that N has a lower Edis than C in Ni and bcc Fe.35–37)
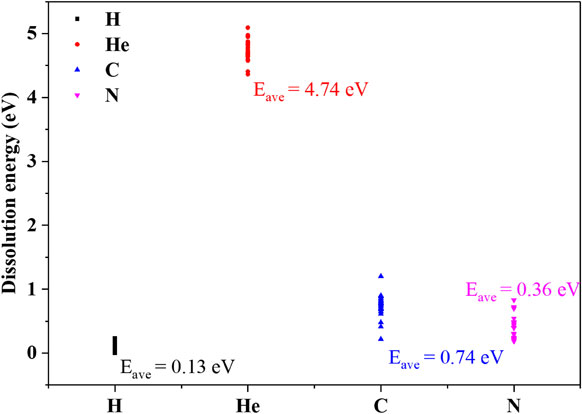
The dissolution energy of H, He, C and N in CrCoFeNi HEA.
Furthermore, it can be seen that the Edis H, He, C and N shows different variety. The variation in Edis of H is small. However, the variation in Edis of He, C and N is quite large. It implies that the dissolution behavior of H at different sites is similar, while the dissolution behavior of He, C and N may strongly depend on the local configuration, for example, the distribution of solute (Cr, Co, Fe and Ni) atoms.
To investigate the relation between the dissolution behavior of interstitial atom and the local distribution of solute (Cr, Co, Fe and Ni) atoms. For each interstitial, 5 configurations with high Edis of interstitial and 5 configurations with low Edis of interstitial are picked, then the number of solute atoms located at the first coordination shell of the interstitial atom (nearest-neighbor, or NN) are counted. Results are shown in Fig. 4. Apparently, H, He and N has low Edis at the site with more Cr atoms as shown in Fig. 4(a) (b) and (d), indicating that H, He and N are more stable at the site with high Cr concentration. However, it seems C has low Edis at the site with less Ni atoms (Fig. 4(c)). The result suggests that an attractive interaction may exist between Cr and H, He, N interstitials. The attractive interaction between Cr and interstitial is considered to be due to the reactive chemical property of Cr. The DFT calculation data of Cr related compounds in Materials Project database38) is investigated. The formation energy of Cr related compounds, Cr3C2, CrH2 and Cr3N2, always has a negative value, while the formation energy of Co, Fe, Ni related compounds, such as Fe3C, Co2C and NiC, has a positive value. It means that Cr containing compounds can be easily formed, which also reveals the high attraction effect of Cr to H, C and N. The attractive interaction between Cr and interstitial was also reported in previous study.34,39,40) However, in this work, no obvious attraction between C and Cr is found. Instead, it seems there is a strong repulsive interaction between C and Ni. After investigating the formation energy of Ni related compounds, it is found that the formation energy of Ni–C compounds is very high. For example, the formation energy of NiC is very high with 1.10 eV, while the formation energy of Fe3C is only 0.053 eV. It reveals that the repulsion effect of Ni to C is strong. Also, previous study reported that there is repulsive interaction between C and Ni.41)
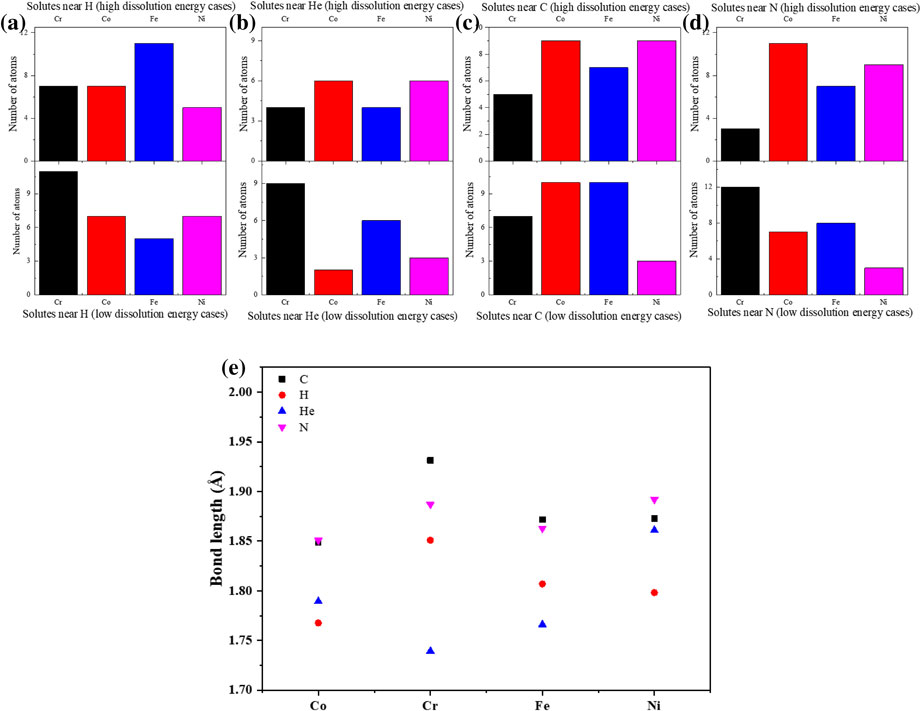
The number of solutes (Cr, Co, Fe and Ni) located at the first coordination shell of H (a), He (b), C (c) and He (d), and the average bond length between interstitial atoms and solutes (e).
Moreover, to get a more complete understanding on the interaction between interstitial and solutes, the bond length of interstitial-solute is investigated, as shown in Fig. 4(e). The bond length of C-solute and N-solute are longer than that of H-solute and He-solute, which is considered to be due to the large size of C and N. It is found that the bond length of He-solute is the shortest, which may be mainly due to He occupies T site but other interstitials occupy O site. The bond length of H-solute, C-solute and N-solute show a similar tendency. Bond with Co has a shorter length, while bond with Cr has a longer length. However, for He, He–Cr bond has the shortest length, while He–Ni bond has the longest length. It implies that the nature of the interaction between H, C, N and the solutes may be very different from the interaction between He and the solutes.
3.3 Effect of H, He, C and N on the physical properties of CrCoFeNi HEAThe effect of H, He, C and N on the magnetic properties of solutes in CrCoFeNi HEA are investigated. The magnetic moment of Cr, Co, Fe and Ni located at the first coordination shell of the interstitial atoms (nearest-neighbor, or NN) is presented in Fig. 5. Although different interstitial atom shows the different effect on magnetic properties of solutes, all of them have the effect of decreasing magnetic moment of Cr, Co, Fe and Ni. Among Cr, Co, Fe and Ni, Fe is the element which is the most affected by interstitial, an obvious decreasing in magnetic moment is observed in Fig. 5(a). While Ni is the least affected one, the decreasing in magnetic moment of Ni near interstitial is within 0.1 µ in average as shown in Fig. 5(d).
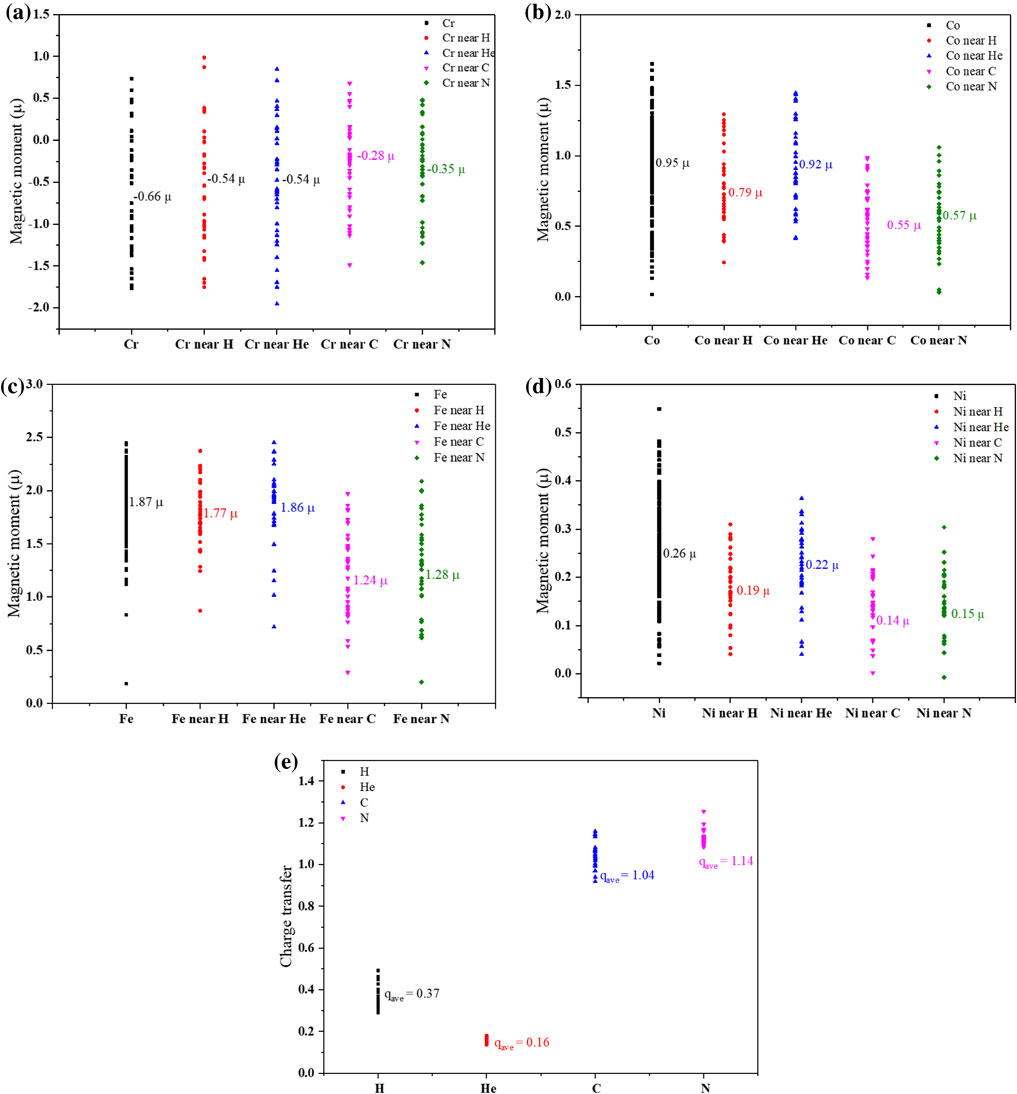
The magnetic moment of Cr (a), Co (b), Fe (c) and Ni (d) next to interstitial in CrCoFeNi HEA and the charge transfer of interstitial atoms (e).
Compared with C and N, the effect of H and He on the magnetic moment of elements in CrCoFeNi HEA is weak. It is considered that the magnetic properties of atoms are strongly related to the charge transfer between atoms since it results in the changing in the unpaired spins of atoms. So the charge transfer of H, He, C and N are investigated as shown in Fig. 5(e). It can be seen that due to the closed-shell structure of He, the electron transferred to He is very limited, only 0.16 electrons in average. It indicates only weak interaction exists between He and the elements in CrCoFeNi HEA. And, consequently, He has little influences on the magnetic properties of solutes. Compared with He, electrons transferred to H is 0.37 electrons in average, which is also low but higher than the cases of He. Therefore, H has a stronger influence on the magnetic moment than He. Electrons transferred to C and N is much higher with 1.04 electrons and 1.14 electrons in average, respectively. It results in the strong effect of C and N on decreasing the magnetic moment of Cr, Co, Fe and Ni.
Last, the electronic structures of HEA with H, He, C and N are investigated. Figure 6 shows the projected density of states (PDOS) of the solutes and the interstitial in HEA. The dot line in Fig. 6 represents the Fermi level. Figure 6(a) shows the PDOS of Cr, Co, Fe and Ni of d-state in HEA without interstitial. The calculated PDOS of Cr, Co, Fe and Ni agree with the former report,42) the major peaks in the PDOS are located above the Fermi level for Cr and below the Fermi level for Co, Fe and Ni. It proves that Cr is more chemically reactive than the other three solutes in CrCoFeNi HEA.
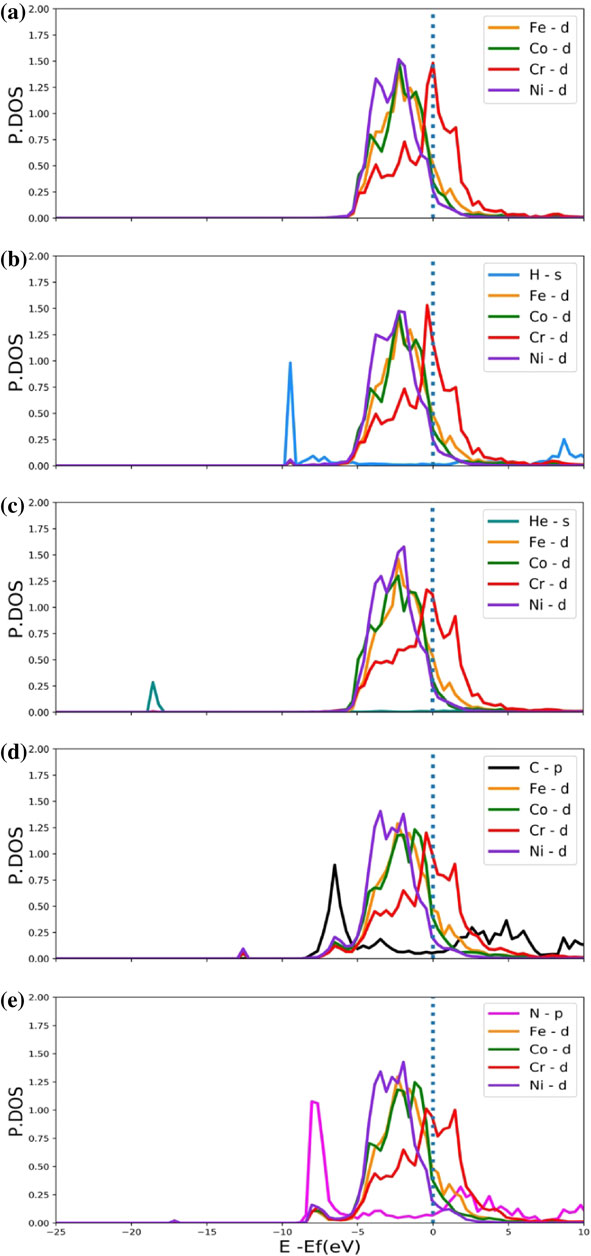
The projected density of states of Cr, Co, Fe and Ni of d-state in HEA without interstitial (a), s-state of H (b) and He (c), p-state of C (d) and N (e), and the nearest Cr, Co, Fe and Ni of d-state.
In Fig. 6(b), it can be seen that at the position of −9 eV, the PDOS of H overlapped with the PDOS of Cr, Co, Fe and Ni which are nearest-neighbor of H, indicating H does slightly interact with Cr, Co, Fe and Ni. This weak interaction is also shown in charge transfer since only 0.37 electrons transferred to H in average. The PDOS of He doesn’t overlap with any solutes as shown in Fig. 6(c). It means no chemical interaction between He and Cr, Co, Fe and Ni, which is mainly due to the closed-shell structure of He. Compared with H and He, the interaction between C, N and Cr, Co, Fe and Ni is much stronger. Figure 6(d) and Fig. 6(e) show that strong hybridization between C, N and Cr, Co, Fe and Ni exists. Moreover, the PDOS of N overlaps with PDOS of Cr, Co, Fe and Ni in the broader region than the PDOS of C, indicating the interaction between N and Cr, Co, Fe and Ni is slightly stronger than the interaction between C and Cr, Co, Fe and Ni.
The dissolution behavior of H, He, C and N in CrCoFeNi high entropy alloys (HEAs) is studied with density functional theory calculation method. The site preference of H, C and N in CrCoFeNi HEAs is the octahedral site, while the site preference of He is the tetrahedral site. The dissolution energy of H is the lowest and it of He is the highest. The high dissolution energy of He is considered to be due to the closed-shell electronic structure of He. Moreover, H, He and N is found to be more stable at the position with more Cr atoms, while C is found to be more stable at the position with less Ni atoms. Furthermore, the effect of H, He, C and N on the physical properties of CrCoFeNi HEAs is studied. H, He, C and N have the effect of decreasing the magnetic moments of solutes in CrCoFeNi HEAs. Electronic structures analysis shows that, for C and N, there is hybridization between C, N and solutes. It implies the chemical bonding between C, N and solutes is strong.
This work was supported by China Scholarship Council. And this work was partly achieved through the use of the supercomputer system at the information initiative center, Hokkaido University, Sapporo, Japan.