Abstract
In this work, a series of cobalt-doped ceria nanorods have been synthesized coming from two cobalt precursors by the impregnation method, based on ceria nanorods pre-formed by the hydrothermal method. The properties of obtained catalysts were investigated by various techniques, including Brunauer-Emmett-Teller nitrogen physisorption measurements (BET), X-ray powder diffraction (XRD), hydrogen temperature-programmed reduction (H2-TPR), scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS), and transmission electron microscopy (TEM). The catalytic activities of as-prepared samples were studied in the deep oxidation of p-xylene at low temperatures (225–300°C). The catalyst characterizations evidenced the crystalline phase formations of CeO2 and Co3O4 with the average crystallite sizes of 17.5–45.8 nm and 11.1–23.4 nm, respectively. The cobalt addition by cobalt nitrate into CeO2 decreased the surface area of CeO2 nanorods (67.9 m2.g−1), in contrast to the increase using cobalt acetate (76.0–82.5 m2.g−1). Co3O4/CeO2 catalysts showed reduction peaks at much lower temperatures than that of pure nanorod ceria. 7.5 mass% Co3O4 supported on nanorod CeO2 catalyst synthesized from cobalt acetate as a type of cobalt precursor with the smallest nanoparticle size and high BET surface area was the most efficient for p-xylene deep oxidation, achieving more than 95% of p-xylene conversion to CO2 at 275°C, and its performance was stabilized for more than 100 hours tested.

1. Introduction
Up to now, many ways to remove VOCs have been reported including absorption, adsorption, thermal incineration, and catalytic deep oxidation.1) Among them, catalytic deep oxidation is well-recognized as one of the most favorable solutions due to its high catalytic performance and moderate operating costs.2,3) In addition, this method permits to remove VOCs perfectly, even at a very dilute concentration and much lower treatment temperature than thermal incineration.
The majority of catalysts for oxidations of VOCs have been studied and applied on the basis of precious metals, mainly Pt, Pd, Ru, and Au. They are highly active at low reaction temperatures, besides major disadvantages such as expensive, easily poisoned by the steam and sintered during reaction at high temperatures. Therefore, the current efforts have devoted d-transition metal oxide catalysts for VOCs removal instead of the traditional ones. Recently, numerous catalytic systems for VOCs oxidation have been reported such as oxidation of 2-propanol over M-Pt/γ-Al2O3 (M = Cr, Mn and Fe);3) toluene over Pt/CeO2,4) nanostructured CeO2–Al2O3,5) semiconductive α-Fe2O36) and catalyst system based upon heater-integrated catalyst units using NiO or Cr2O3;7) C9 to C10 paraffins and napthtenics on CuO/Al2O3;8) m-xylene on CeO2–Al2O3;5) p-xylene on CuO–CeO2/ZRM;9) etc. Frequently, metal oxides based on Cu, Cr, Mn, Fe, Co, and Ni demonstrate high catalytic activity in the deep oxidation. Prasad et al.10) reported that in the oxidation of CO, their catalytic activity decreases in the order: Co3O4 > CuO > MnO2 > Fe2O3 > Cr2O3 > NiO. In particular, the activity of Co3O4 catalyst is approximately equal to that of Pt-based catalysts, but its catalytic activity and stability need to be further improved. From an industrial application point of view, the use of supported Co3O4 nanoparticles is still preferred rather than pure Co3O4 nanomaterials because of their higher activation and sintering resistance.11)
The most frequently used method is probably aqueous impregnation of various supports with cobalt salts. In order to investigate the effect of the metal dispersion in cobalt catalysts, alternative impregnation preparations using of various types of cobalt precursors involved cobalt nitrate,12) cobalt acetate,13) or organic chelating agents with a suitable stoichiometry. Beyond the metal dispersion, the reduction potential of the active cobalt species, in particular that of the Co(III)/Co(II) redox couple, is also important factor that may affect the oxidation activity of the catalyst.14,15) The reducing ability is generally dependent on several variables such as the type of Co precursor, the type of promoter and its loading, and the type of support.16) However, studies on the effect of cobalt precursors on the oxidative activity of Co-based catalysts are limited.
Add another insight about cobalt catalyst,17) Co3O4 supported on Al2O3 catalyst performed the high activity for CO oxidation at 21°C, and the activity of catalysts based on cobalt oxide depends on its phase states. Only Co3O4 phase showed high activity but it is sensitive and heat-unstable, provoking the further studies on suitable preparation and alternative supports. Mei et al.18,19) showed that Co3O4 catalysts supported on TiO2 and CeO2 were highly efficient in the oxidation of dibromomethane. CeO2 is often used as a support in the catalytic reaction, due to its high oxygen storage capacity, prominent oxygen mobility, and some acidic properties. Besides, CeO2 support can increase the metal-support interaction and promote the dispersion of metal species. In fact, Co3O4/CeO2 catalysts have been studied in the field of catalysis due to their unique physicochemical properties and the catalytic performance of CeO2 is affected by exposed lattice planes.19) In general, different lattice planes are exposed in different CeO2 morphologies, for instance, CeO2 nanorods are prone to expose two (100) planes and four (110) planes,20) while CeO2 nanocubes expose six (100) planes.21) The high catalytic activity of Co supported on CeO2-rod was attributed to the high content of Co3+, more surface-adsorbed oxygen, and more oxygen vacancies related to their exposed (100) and (110) planes.19) In addition, Co3O4 has a strong interaction with CeO2-rod resulting in the superior and long-duration stability compared to that of other CeO2 morphologies in oxidation processes.
To have more insight of Co3O4–CeO2 catalysts, in this study, Co3O4-doped CeO2 nanorods were prepared by the impregnation method. The effects of precursor and cobalt loading on physicochemical properties and catalytic behavior as well as stability of cobalt-doped ceria nanorods in p-xylene deep oxidation were investigated.
2. Experimental
Unless otherwise stated, all the chemicals are purchased from Merck and used directly without further purification. Nanorod CeO2 was synthesized using the hydrothermal method reported previously.22) Firstly, 2.17 grams of Ce(NO3)3.6H2O and 15.40 grams of NaOH were dissolved separately in 20 mL and 55 mL of distilled water, respectively. Then, NaOH solution was dropped continuously into Ce(NO3)3 solution and the mixture was kept stirring at 600 rpm within 1 hour before being transferred into a Teflon bottle (100 mL). A Teflon bottle was packed in a tightly sealed autoclave and hydrothermally treated at 130°C for 5 hours. After that, the resulting precipitate was separated and washed thoroughly with distilled water to remove Na+ (until pH became 7). The precipitate was collected by centrifugation and dried at 80, 100, and 120°C for 2 hours at each temperature, and finally calcined at 500°C for 2 hours in the air with the heating rate of 5°C.min−1 and the airflow rate of 6 L.h−1 to obtain nanorod CeO2.
Co3O4 doped on nanorod CeO2 catalyst was prepared by impregnating cobalt salt solution on nanorod CeO2. After the wet impregnation, the obtained suspension was kept at room temperature overnight and drying in air at 80°C, 100°C, and 120°C within 2 hours at each temperature, and then the sample was calcined at 350°C for 1 hour in the airflow of 6 L.h−1.
To survey the effect of precursor on the catalytic behavior of Co3O4 doped on nanorod CeO2, catalysts 5 mass% Co3O4/CeO2 nanorods were prepared from two cobalt salts including cobalt nitrate (Co(NO3)2.6H2O) and cobalt acetate (Co(OAc)2.4H2O). Based on this result, a suitable precursor for the synthesis of Co3O4/CeO2 nanorods was proposed. Next, to survey the effect of cobalt loading on the behavior of catalysts, the Co3O4 content in the catalysts was changed, being 5.0; 7.5; and 10.0 mass% using the precursor solution of suitable cobalt salt, and pure CeO2 was also used as a control sample. The catalysts of Co3O4 doped on nanorod CeO2 are symbolized as following xCo(y)-rCe, representing Co3O4 content of x mass% loaded onto CeO2 nanorods (rCe) derived from cobalt nitrate (y = N) or cobalt acetate (y = A).
X-ray powder diffraction (XRD) patterns were collected on Bruker D2 Phaser diffractometer using CuKα radiation (λ = 0.154 nm) in 2θ = 20–80°. The average crystal size of CeO2 or Co3O4, d (nm), was calculated according to eq. (1).23)
| \begin{equation}
d(\textit{nm}) = \frac{K\lambda}{\beta \cos \theta},
\end{equation}
| (1) |
where K, the Scherrer constant, is taken to be 0.94, λ is the wavelength of the X-ray, β is the line width at half maximum height of the peak in radians, and θ is the position of the peak in radians. The surface area, pore volume, and average pore size of the catalysts were measured by nitrogen adsorption-desorption isotherm at −196°C on Nova Station B, Quanta chrome Nova Win Instrument. Samples were degassed in vacuum at 200°C for 2 hours before analyzing. The morphology of catalysts was characterized by transmission electron microscopy (TEM) using a JEOL JEM 1400 instrument with an acceleration potential of 100 kV. Samples were put into ethanol, sonicated for thirty minutes and dropped onto copper grids to make a TEM sample. The hydrogen temperature-programmed reduction (H
2-TPR) measurement was performed on Gas Chromatograph GOWMAC 69-350 with a thermal conductivity detector (TCD). 50 miligrams of sample was reduced
in-situ with a flow of 10%H
2/N
2 with a total gas flow rate of 30 mL.min
−1. The temperature of the reactor was raised linearly from 50 to 900°C at a heating rate of 10°C.min
−1. Samples were analyzed by XPS analysis using a Kratos SUPRA XPS fitted with a monochromated Al kα X-ray source (1486.7 eV, 15 kV, and 10 mA).
The catalytic behavior of samples expressed as the conversion of p-xylene was measured in a fixed tubular quartz reactor (8 mm i.d.) under atmospheric pressure under atmospheric pressure at a temperature range of 225 to 300°C. The weight hourly space velocity (WHSV) was 60,000 mL.h−1.g−1, and the mass of catalyst was 0.2 grams with a size range of 0.25 to 0.50 mm. Before testing the activity, the sample was activated at 300°C for 1 hour in airflow with a velocity of 30,000 mL.h−1.g−1. The inlet concentrations of p-xylene and oxygen in nitrogen were 0.34 and 10.5 mol%, respectively. The inlet and outlet mixtures were analyzed with an Agilent 6890 Plus Gas Chromatograph with a FID detector using capillary column DB-624 and with a TCD detector using capillary column TG-BONQ. The tests were conducted in triplicate to ensure the accuracy of the results. To ensure accuracy of the results, the tests were undertaken in triplicate. The stability of the best catalyst was investigated under the reaction temperature of 275°C in 100 hours.
3. Results and Discussions
3.1 Catalyst characteristics
The XRD patterns of the pure nanorod CeO2 (rCe) and Co3O4/CeO2 (Co-rCe) catalysts are shown in Fig. 1. The XRD pattern of pure CeO2 nanorods is in agreement with a face-centered cubic CeO2 phase (JCPDS-34-0394).24,25) CeO2 crystal structure did not change after the Co3O4 loading. Besides, there are two small peaks at 2θ = 36.4 and 66.1° corresponding to the (311) and (440) planes of Co3O4, respectively. Diffraction peaks of Co3O4 species were observed only in the samples of 7.5Co(A)-rCe and 10Co(A)-rCe, where Co3O4 concentration was loaded up to 7.5–10 mass%. Based on the XRD highest peak of at 2θ = 28.7° and 36.4°, the average crystal size of CeO2 and Co3O4 was determined following the equation the Scherrer equation (1) shown in Table 1. All samples have high degree of crystallinity with small-sized particles of CeO2 (17.5–45.8 nm) and Co3O4 (11.1–23.4 nm). Among them, the particle size of 7.5Co(A)-rCe was the smallest.

Table 1 Textural characteristics of the prepared samples.
The main textural and structural characteristics of ceria nanorods doped and undoped Co3O4 are summarized in Table 1. The pure ceria sample (CeO2-NR) exhibited a BET value of 71.8 m2.g−1, a pore volume of 0.343 cm3.g−1 and an average pore diameter of 27.5 Å. The addition of Co3O4 derived from cobalt nitrate into CeO2 decreased the surface area of CeO2 nanorods (67.9 m2.g−1), but it increased in the case of using cobalt acetate as a type of cobalt precursor (76.0–82.5 m2.g−1). Regarding the average pore diameter and pore volume, both types decreased upon the addition of Co to ceria nanorods. In which, the addition of cobalt leads to a slight decrease in the average pore diameter (22.4–23.6 Å), whereas the pore volume is significantly dropped (0.080–0.098 cm3.g−1). Besides, it can be seen that the BET surface area, pore diameter and pore volume of Co(A)-rCe catalysts decreased as the Co3O4 loading amount increased on the surface of CeO2 nanorods. The results of the adsorption-desorption isotherms of nanorod CeO2 catalysts undoped and doped Co3O4 in Fig. 2 show that all samples obey type IV isotherms with a hysteresis loop at a relative pressure >0.5, further corroborating the mesoporous structure of the catalysts.26,27) Figure 3 shows the BJH of ceria nanorods and Co-rCe catalysts. According to the pore size distribution, all the samples have their maxima at a pore diameter of more than 10 nm, designating the presence of mesopores.28) It was evident that ceria nanorods and Co-rCe samples exhibited nearly similar particle size distributions. It demonstrated that pore size distributions remain practically unaffected by the addition of cobalt in all cases.


The oxidation state of catalyst surface species is examined by XPS analysis (Fig. 4). The 7.5Co(A)-rCe sample was chosen as the representative for the following discussion. Figure 4(a) represented the XPS patterns for Ce 3d core level of 7.5Co(A)-rCe. Ce appeared to be just in the form of pure Ce4+ with six peaks labeled as V0 (881.9 eV), V1 (888.5 eV), V2 (897.8 eV), V0′ (900.8 eV), V1′ (907.3 eV) and V2′ (916.2 eV).29) The high binding energy (BE) doublet (V2/V2′) is attributed to the final state of Ce(IV) 3d94f0O2p6, doublet V1/V1′ is originated from the state of Ce(IV) 3d94f1O2p5, and doublet V0/V0′ corresponds to the state of Ce(IV) 3d94f2O2p4. The characteristic peaks of Ce3+ were not almost observed. Therefore, Ce species exist mainly in the tetravalent oxidation state (Ce4+ ions).30) Figure 4(b) exhibits the Co 2p XPS pattern of 7.5Co(A)-rCe sample. The sample exhibited a major peak of Co2p3/2 (780 eV).31) The Co2+/Co3+ ratio of 7.5Co(A)-rCe sample reached at 2.54 (Table 2). So, Co3O4 (CoO.Co2O3) is considered as the main phase in pure cobalt oxide in agreement with XRD result. The O 1s spectra of 7.5Co(A)-rCe (Fig. 4(c)) show the peaks at high (529.0 eV) and medium (531.0 eV) binding energies indicating lattice (O2−) and surface adsorbed oxygen species (O2−, O−, O22−) respectively. Besides, a broad shoulder at 533.5 eV refers to the contribution of the defective oxides or the presence of hydroxylated or carbonated oxygen on the surface of the catalysts. This property confirms the evidence of the enhanced lattice oxygen storage and mobility incurred by the strong interaction of CeO2–Co3O4.32)


Table 2 XPS results for 7.5Co(A)-rCe sample.
Figure 5(a) represents a magnification TEM image of the as-synthesized CeO2 nanorods, which are typically 50–100 nm in length and 5–20 nm in diameter. Figures 5(b)–(e) illustrate the images derived by TEM analysis for the Co-rCe mixed oxides. The crystallite of a spheroidal shape with an internal diameter of approximately 5–20 nm is mainly the Co-containing phase.33) The morphology is not affected by cobalt addition to the nanorod ceria support. It can find that the crystalline spheres of Co-rCe synthesized using cobalt acetate precursor are smaller and more uniformly dispersed than one using nitrate precursor. A comparison of these results with the XRD and BET surface analysis suggested that the use of cobalt acetate precursor for the synthesis of Co3O4 doped CeO2 can increase the dispersion of Co3O4 species and surface area, and reduce nanoparticle size of the catalyst.

Figure 6 shows that the H2-TPR profile of nanorod CeO2 appears two reduction peaks at 460 and 480°C for the surface and bulk reductions, respectively. It also exhibits a broad peak above 800°C, attributed to the ceria subsurface oxygen reduction.34) Co3O4 doped CeO2 catalysts show reduction peaks in much lower temperatures than that of ceria nanorod. It can be found that the Co3O4–CeO2 catalysts appeared three reduction peaks. The first peak at temperature range of 200–320°C is contributed by the reduction of Co3+ to Co2+ and/or Co2+ to Co0, the second peak with low intensity at temperature range of 350–450°C was attributed to characteristic for Co3O4 bound to CeO2, and the third broad one at 750–850°C could be associated to the ceria subsurface oxygen reduction.35,36) The 7.5Co(A)-rCe sample shifted to the lower temperature range with the highest reduction peaks, therefore this catalyst was reduced more easily corresponding to the highest hydrogen consumption. This proved that Co3O4 particles have better dispersion with smaller size on this sample.

The catalytic activity of the as-prepared materials in p-xylene oxidation was shown in Fig. 7. The analysis result of gas mixtures in the output of the reactor showed that CO2, H2O, N2, O2, and remain p-xylene were only detected. It proved that p-xylene was oxidized deeply to CO2 and H2O on all surveyed catalysts. The effect of cobalt precursor on the activity in the oxidation of catalysts was first examined. Figure 7(a) showed the p-xylene conversion increased with a high-level temperature. Noticeably, the completely conversion of p-xylene was obtained at ∼300°C on both catalysts. The effect of CeO2 could be ascribed that CeO2 in contact with Co3+ facilitates the formation of Co2+ due to its redox process of Ce4+/Ce3+ which could transfer electrons to Co3+ resulting in formation of Co2+,37) thus enhancing the adsorption of p-xylene. The formation of Co3O4–CeO2 interfacial sites produced the synergistic redox properties.38) Besides, CeO2 also serves as an oxygen supplier through either the formation of superoxide species (O2−) by gas phase oxygen reacting with oxygen vacancies on its surface or the direct involvement of lattice oxygen.39) Furthermore, the oxygen vacancies are easily formed in the CeO2 support thanks to its high oxygen mobility to improve the catalytic activity. Therefore, oxygen vacancies play an important role in the reaction since they provide sites for oxygen activation to form superoxide (O2−), which was considered as the intermediate species in the oxidative reactions occurring on the catalyst surface. Therefore, the catalysts are highly active during the oxidation of p-xylene. Obviously, the 5Co(A)-rCe sample has better catalytic activity than that of 5Co(N)-rCe at a range temperature at 225–275°C. This result is closely related to the obtained characteristics of 5Co(A)-rCe and 5Co(N)-rCe catalysts including specific surface area (BET), Co3O4 crystal size (XRD and TEM) as well as reduction properties (H2-TPR). So, the cobalt acetate as a suitable cobalt precursor for synthesizing Co3O4/CeO2 catalyst was chosen for further surveys.
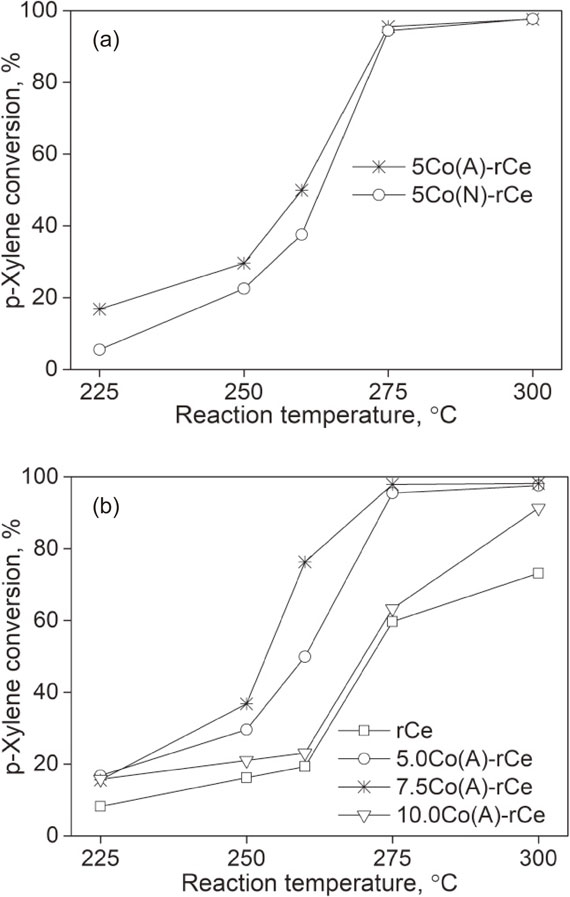
Effect of cobalt loading derived from cobalt acetate precursor on the activity in p-xylene oxidation of catalysts is also investigated and the obtained results are shown as in Fig. 7(b). Apparently, the addition of cobalt in the ceria lattice enormously enhances the catalytic efficiency compared to pure ceria nanorods sample. For all Co3O4/CeO2 samples, conversion of p-xylene dramatically raised when the reaction temperature increased from 225 to 300°C. Generally, it reached over 85% at 400°C. The 7.5Co(A)-rCe sample was the most efficient, as the p-xylene conversion reached 95% of p-xylene at a reaction temperature of 275°C and the full conversion at 300°C. This is quite consistent with its H2-TPR result. Co3O4 and CeO2 phases on 7.5Co(A)-rCe were reduced more easily compared to others (Fig. 6). Balzer et al.40) synthesized Co2O3 catalysts supported on CeO2 + γ-Al2O3 for the deep oxidation of VOCs including n-hexane, benzene, toluene and o-xylene. The sample with 20 mass% Co2O3 was the best one for such reactions, and conversion of n-hexane, benzene, toluene and o-xylene reached 96, 90, 70 and 58%, respectively at reaction temperature of 350°C. Meanwhile, 20 mass% Co2O3/γ-Al2O3 undoped CeO2 just reached 50, 45, 35 and 25% under same conditions. Wu et al.41) prepared Au catalyst loaded (1.5 mass%) on the different supports (ZnO, Al2O3, and MgO) by a colloidal deposition method for the oxidation of p-xylene. The p-xylene conversion achieved 30, 14 and 9% at reaction temperature of 300°C on Au/ZnO, Au/Al2O3 and Au/MgO catalysts, respectively. In comparison to the activity of these catalysts as mentioned, 7.5Co(A)-rCe catalyst, a highly active catalyst oxidizing over 95% of p-xylene to CO2 at 275°C.
The most outstanding catalyst among those tested was selected for the time-stability test. Accordingly, the stability of 7.5Co(A)-rCe catalyst was investigated at 275°C during 100 hours, the conversion of p-xylene is recorded by time. The obtained results in Fig. 8 indicate the reality of expectation about the catalyst. As can be observed, the p-xylene conversion was kept almost stable during 100 hours tested (Xp-xylene = 93–96%). The above results can be seen that 7.5Co(A)-rCe catalyst is both highly reactive and stable in the deep oxidation of p-xylene. The XRD and TEM results of the catalysts (Fig. 5, 9 and 10) showed that the phase composition as well as the morphology of the spent 7.5Co(A)-rCe catalyst was almost unchanged compared to the fresh one. This proves that the structure of the catalyst is not damaged under tested reaction conditions at 275°C after 100 hours of time on stream. These results showed the potential application of this catalyst in gas-phase VOCs treatment.
4. Conclusion
Deep catalytic oxidation of p-xylene has been conducted over both pure CeO2 and Co3O4 doped CeO2 nanorods. When using cobalt acetate as a type of the cobalt precursor, the insertion of cobalt ions in CeO2 lattice and the decrease of Co3O4 crystal size resulted in an increasing of surface area, a decreasing of the reducing temperature, and a rising of the available lattice oxygen in bulk. These properties play a vital role in the superior catalytic activity of Co3O4 doped CeO2. The suitable cobalt loading was determined. Most promisingly, the 7.5Co(A)-rCe sample was the most active one, as the full conversion of p-xylene to CO2 and H2O at the reaction temperature of 300°C. This work opens an alternative way for the further application of Co3O4 doped nanorod CeO2 in VOCs deep oxidation.
Acknowledgments
The study was supported by The Youth Incubator for Science and Technology Programme, managed by Youth Development Science and Technology Center - Ho Chi Minh Communist Youth Union and Department of Science and Technology of Ho Chi Minh City, the contract number is “03/2019/HĐ-KHCNT-VƯ”.
REFERENCES
- 1) Y.M. Salih, A.R. Karim and I. Khurshid: Petrol. Sci. Technol. 36 (2018) 1037–1043.
- 2) H.S. Kim, T.W. Kim, H.L. Koh, S.H. Lee and B.R. Min: Appl. Catal. A Gen. 280 (2005) 125–131.
- 3) A. Niaei, D. Salari, F. Aghazadeh, N. Caylak and A. Sepehrianazar: J. Environ. Sci. Health A 45 (2010) 454–463.
- 4) R. Peng, S. Li, X. Sun, Q. Ren, L. Chen, M. Fu, J. Wu and D. Ye: Appl. Catal. B 220 (2018) 462–470.
- 5) L.M. Dai, D.N. Nhiem, D.T. Lim and N.D. Van: Mater. Trans. 54 (2013) 1060–1062.
- 6) S. Ito, Y. Yui and J. Mizuguchi: Mater. Trans. 51 (2010) 1163–1167.
- 7) A. Maki and J. Mizuguchi: Mater. Trans. 51 (2010) 1361–1363.
- 8) K. Tzortzatou and E. Grigoropoulou: J. Environ. Sci. Health A 45 (2010) 534–541.
- 9) N. Tinh, N. Van, N. Anh, H. Ha and N. Tri: J. Environ. Sci. Health A 54 (2019) 352–358.
- 10) R. Prasad and P. Singh: Catal. Rev. 54 (2012) 224–279.
- 11) A.Y. Khodakov, W. Chu and P. Fongarland: Chem. Rev. 107 (2007) 1692–1744.
- 12) J. Dou, Y. Tang, L. Nie, C.M. Andolina, X. Zhang, S. House, Y. Li, J. Yang and F.F. Tao: Catal. Today 311 (2018) 48–55.
- 13) L. Wei, X. Dong, M. Ma, Y. Lu, D. Wang, S. Zhang, D. Zhao and Q. Wang: Int. J. Hydrogen Energy 43 (2018) 1529–1533.
- 14) Z. Fan, W. Fang, Z. Zhang, M. Chen and W. Shangguan: Catal. Commun. 103 (2018) 10–14.
- 15) H. Wang, D. Mao, J. Qi, Q. Zhang, X. Ma, S. Song, L. Gu, R. Yu and D. Wang: Adv. Funct. Mater. 29 (2019) 1806588.
- 16) J. Waikar, H. Pawar and P. More: Catal. Green Chem. Eng. 2 (2019) 11–24.
- 17) J. Jansson: J. Catal. 194 (2000) 55–60.
- 18) J. Mei, S. Zhao, H. Xu, Z. Qu and N. Yan, RSC Adv. 6 (2016) 31181–31190.
- 19) J. Mei, Y. Ke, Z. Yu, X. Hu, Z. Qu and N. Yan: Chem. Eng. J. 320 (2017) 124–134.
- 20) X. Du, D. Zhang, L. Shi, R. Gao and J. Zhang: J. Phys. Chem. C 116 (2012) 10009–10016.
- 21) Z. Liu, S. Guo, C. Hong and Z. Xia: J. Mater. Sci. Mater. Electron. 27 (2016) 2146–2150.
- 22) L.C. Loc, P.H. Phuong, D. Putthea, N. Tri, N.T.T. Van and H.T. Cuong: Int. J. Nanotechnol. 15 (2018) 968–982.
- 23) A.L. Patterson: Phys. Rev. 56 (1939) 978–982.
- 24) D.M. Walker, S.L. Pettit, J.T. Wolan and J.N. Kuhn: Appl. Catal. A Gen. 445 (2012) 61–68.
- 25) H. Ay and D. Üner: Appl. Catal. B 179 (2015) 128–138.
- 26) L. Wang, H. Liu, Y. Chen and S. Yang: Int. J. Hydrogen Energy 42 (2017) 3682–3689.
- 27) L. Tan, Q. Tao, H. Gao, J. Li, D. Jia and M. Yang: J. Porous Mater. 24 (2017) 795–803.
- 28) A. Allwar, M. Noor and M. Nawi: J. Phys. Sci. 19 (2008) 93–104.
- 29) Y. Liu, C. Ma, Q. Zhang, W. Wang, P. Pan, L. Gu, D. Xu, J. Bao and Z. Dai: Adv. Mater. 31 (2019) 1900062.
- 30) E. Bêche, P. Charvin, D. Perarnau, S. Abanades and G. Flamant: Surf. Interface Anal. 40 (2008) 264–267.
- 31) T. Chen, S. Li, L. Ma, X. Zhao and G. Fang: Nanotechnology 30 (2019) 395403.
- 32) W. Tang, X. Wu, D. Li, Z. Wang, G. Liu, H. Liu and Y. Chen: J. Mater. Chem. A 2 (2014) 2544–2554.
- 33) C.-B. Wang, H.-K. Lin and C.-W. Tang: Catal. Lett. 94 (2004) 69–74.
- 34) X. Liu, K. Zhou, L. Wang, B. Wang and Y. Li: J. Am. Chem. Soc. 131 (2009) 3140–3141.
- 35) J.-Y. Luo, M. Meng, X. Li, X.-G. Li, Y.-Q. Zha, T.-D. Hu, Y.-N. Xie and J. Zhang: J. Catal. 254 (2008) 310–324.
- 36) L. Liotta, G. Di Carlo, G. Pantaleo, A. Venezia and G. Deganello: Appl. Catal. B 66 (2006) 217–227.
- 37) S. Lu, F. Wang, C. Chen, F. Huang and K. Li: J. Rare Earths 35 (2017) 867–874.
- 38) J. Mei, Y. Ke, Z. Yu, X. Hu, Z. Qu and N. Yan: Chem. Eng. J. 320 (2017) 124–134.
- 39) T. Montini, M. Melchionna, M. Monai and P. Fornasiero: Chem. Rev. 116 (2016) 5987–6041.
- 40) R. Balzer, L.F.D. Probst, V. Drago, W. Schreiner and H.V. Fajardo: Braz. J. Chem. Eng. 31 (2014) 757–769.
- 41) H. Wu, L. Wang, J. Zhang, Z. Shen and J. Zhao: Catal. Commun. 12 (2011) 859–865.



